Stevens–Johnson syndrome
Stevens–Johnson syndrome (SJS) is a rare, severe mucocutaneous reaction—usually drug-related—characterized by widespread epidermal cell death, blistering, and mucosal erosions that require urgent supportive care.
Overview
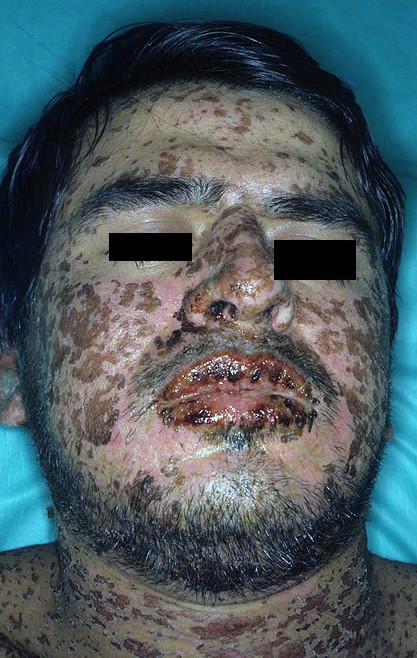
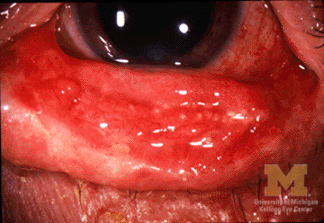
Stevens–Johnson syndrome (SJS) is an uncommon but serious disorder that affects the skin and mucous membranes. It presents with an initial flu-like illness followed by rapidly spreading skin pain, blistering and peeling that resembles a burn. Mucosal surfaces of the mouth, eyes and genitals are frequently involved, which contributes to acute complications and lasting disability. For summaries of clinical features and management see skin and mucous membrane resources.
Image gallery
4 Images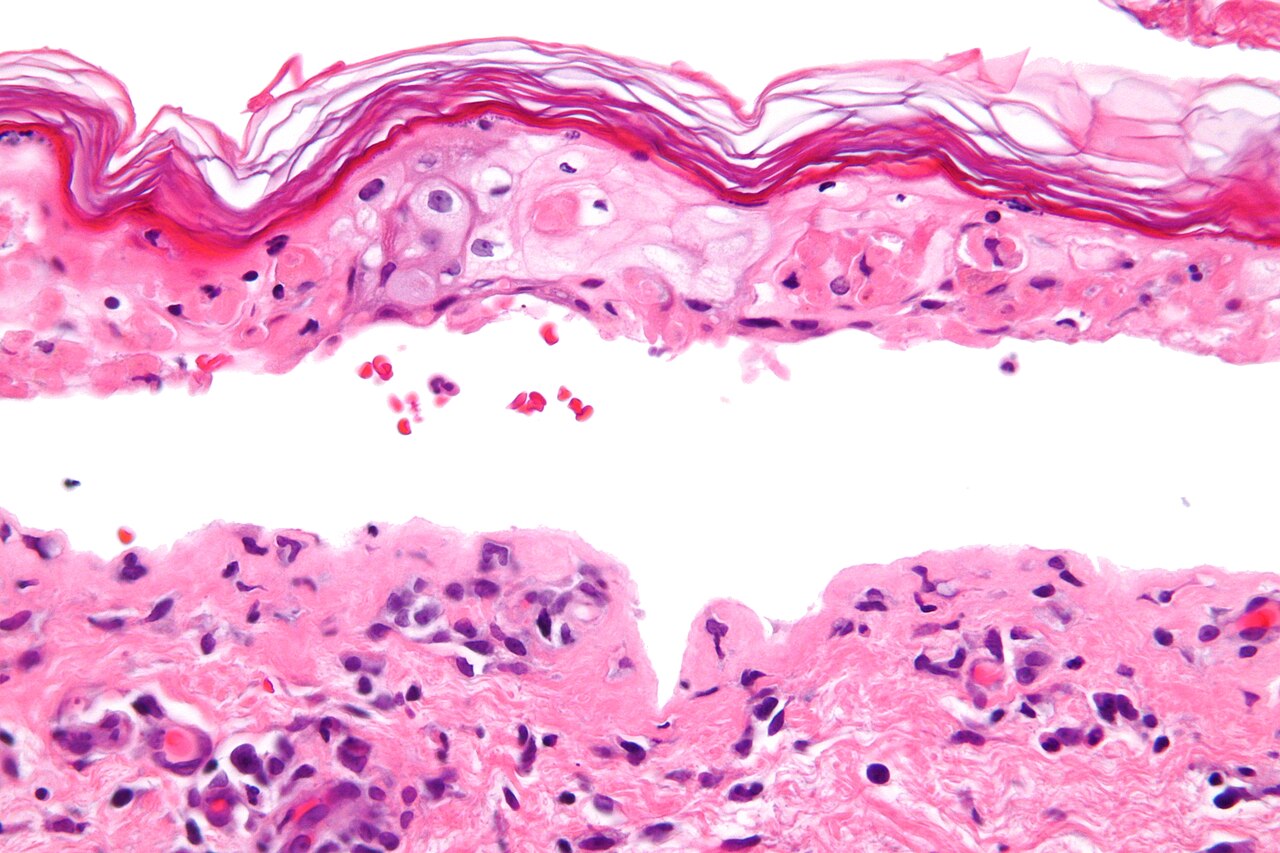

Causes and mechanism
Most cases of SJS are triggered by medications, although infections and, less commonly, underlying malignancy or idiopathic factors can be responsible. The condition reflects an immune-mediated process in which cytotoxic lymphocytes and inflammatory mediators cause widespread keratinocyte death and detachment of the epidermis from the dermis. Detailed discussions of pathogenesis and immune mechanisms can be found in reviews of drug-induced immune reactions.
- Common drug triggers include certain antibiotics, anticonvulsants, allopurinol and some nonsteroidal anti-inflammatory drugs; clinicians and patients should be aware of high-risk culprit drugs.
- Infections such as Mycoplasma pneumoniae and some viral illnesses can precipitate SJS, particularly in children; see information on infectious associations at infections such as Mycoplasma.
- Rarely, SJS occurs in association with cancers (for example lymphomas) or without an identifiable cause; more on those links is available via lymphoma and malignancies.
Clinical features and diagnosis
Patients typically experience a prodrome of fever, sore throat and malaise. Within days they develop painful red or purplish patches that blister and slough, producing raw areas and erosions. Mucosal involvement is common and can lead to difficulty eating, urination problems and severe eye disease. The extent of skin detachment helps classify the disorder and guides prognosis; histologic examination shows full‑thickness epidermal necrosis and minimal inflammation, reflecting the process of epidermal separation described in many dermatology references (epidermal detachment).
Management
Immediate steps include stopping any suspected offending medication and providing care in an environment equipped for large skin wounds (often a burn unit or intensive care setting). Treatment is principally supportive: fluid and electrolyte management, wound care, pain control, nutritional support and prevention or treatment of secondary infection. Specialized eye care is important to reduce the risk of long-term visual loss.
- Supportive measures and wound management are the foundation of therapy; see clinical care guidelines at dermatology resources.
- Several systemic therapies (corticosteroids, intravenous immunoglobulin, cyclosporine and biologic agents) have been used, but evidence varies and choices are individualized; experts recommend multidisciplinary review when considering these options.
Prognosis, complications and prevention
Prognosis depends on the extent of skin involvement, patient age and coexisting illness. Serious short-term risks include fluid loss, infection and organ dysfunction; long-term problems can include scarring, pigment changes and disabling ocular damage. Risk stratification scores are sometimes used in hospitals to estimate mortality risk and guide treatment intensity. Prevention focuses on avoiding known triggers—documenting drug allergies, cautious prescribing, and, in some populations, genetic screening for high‑risk alleles before starting particular drugs. Further reading and patient resources are available at lymphoma and malignancies and skin and mucous membrane resources.
Related articles
Author
AlegsaOnline.com Stevens–Johnson syndrome Leandro Alegsa
URL: https://en.alegsaonline.com/art/93883