Neuroblastoma: a pediatric cancer of the sympathetic nervous system
Neuroblastoma is a childhood cancer arising from neural crest–derived sympathetic cells, most often in the adrenal medulla; presentation, biology, and outcome vary widely with age, stage, and genetics.
Overview
Neuroblastoma is a malignant tumor that arises from immature nerve cells of the sympathetic nervous system. It is primarily a disease of infants and young children and is one of the most common extracranial solid tumors in childhood. Tumors develop from neural crest elements and are most frequently found in the adrenal medulla but can originate anywhere along the sympathetic chain, including the neck, chest, abdomen, and pelvis. The disorder affects the autonomic nervous system and is therefore connected to tissues that regulate involuntary body functions. While neuroblastoma is a cancer, it shows unusual biological diversity: some tumors progress aggressively, while others may stabilize or regress spontaneously.
Image gallery
6 Images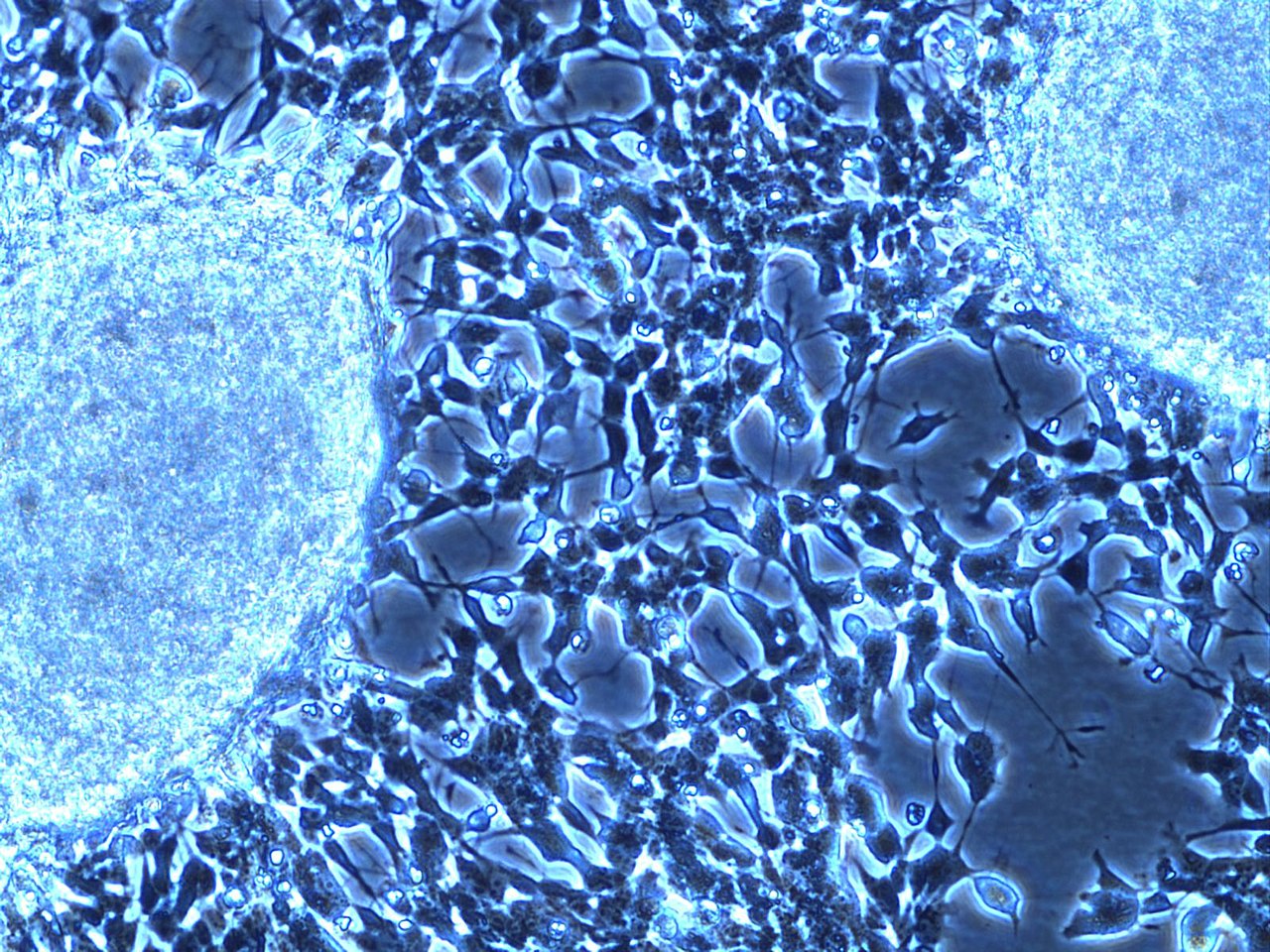



Characteristics and typical presentation
Neuroblastoma cells are poorly differentiated nerve cells that form solid masses. Common signs and symptoms depend on tumor location and spread. In the abdomen, a painless mass or distension is often noticed. Thoracic tumors may cause breathing or swallowing difficulties. When tumors compress spinal nerves, children can develop weakness or paralysis. Because many neuroblastomas produce catecholamines, biochemical signs may include elevated urinary or blood metabolites. Rare paraneoplastic phenomena — such as an involuntary eye movement disorder with myoclonus — can occur.
- Frequent clinical features: abdominal mass, pain, weight loss, bone pain from metastases.
- Possible biochemical clues: elevated catecholamine metabolites (e.g., VMA/HVA).
- Paraneoplastic syndromes: opsoclonus-myoclonus-ataxia and other immune-mediated effects.
Diagnosis and staging
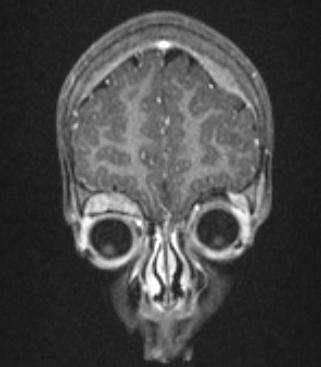
Diagnosis combines imaging, laboratory testing, and tissue sampling. Imaging modalities include ultrasound, computed tomography (CT) and magnetic resonance imaging (MRI) to define tumor extent, and functional imaging such as meta-iodobenzylguanidine (MIBG) scans to detect catecholamine-avid disease. Biopsy or surgical resection provides tissue for microscopic examination; typical histology shows small round blue cells and may display Homer Wright rosettes. Laboratory evaluation often measures catecholamine metabolites. Staging systems and risk stratification take into account age at diagnosis, extent of spread, and tumor biology.
Current clinical assessment incorporates genetic and molecular features into prognosis. Tumor amplification of the MYCN oncogene, certain chromosomal deletions, and DNA ploidy are among the biological markers that influence risk categorization and treatment decisions.
Treatment approaches and prognosis
Management is tailored to the child’s age, disease stage, and tumor biology. Low-risk tumors — particularly small lesions in very young infants — can sometimes be observed because of spontaneous regression. Standard treatments include surgery to remove localized disease, chemotherapy for systemic control, radiation therapy for residual or refractory tumors, and stem cell transplantation for high-risk cases. In recent decades, immunotherapy (for example, anti-GD2 antibodies) and differentiation therapy using retinoids have improved outcomes for some high-risk patients.
- Local control: surgical resection when feasible.
- Systemic therapy: multi-agent chemotherapy and consolidation with autologous stem cell transplant for high-risk disease.
- Adjunct treatments: radiation, anti-GD2 immunotherapy, and differentiation agents.
Prognosis ranges from excellent for many infants with localized disease to guarded for older children with metastatic, biologically aggressive tumors. Late effects of therapy — including growth, endocrine, hearing, and fertility issues — are an important aspect of long-term care.
History, research directions, and notable facts
Neuroblastoma was recognized as a distinct pediatric tumor in the 20th century as neuropathology developed. Research continues to define the molecular drivers that determine tumor behavior and to translate those findings into targeted therapies. Notable features of neuroblastoma include its origin in embryonal neural crest cells and the rare phenomenon of spontaneous regression, especially in infants with specialized low-burden presentations sometimes called stage 4S. Genetic markers such as MYCN amplification are strongly associated with poor outcome and are central to risk-adapted treatment strategies.
Importance and resources
Because neuroblastoma affects very young children and can behave unpredictably, prompt evaluation by pediatric oncology teams is critical. Multidisciplinary care coordinates surgery, medical therapy, radiation, and supportive services. For further information on clinical guidelines, research updates, and family support, consult authoritative sources and professional organizations that specialize in pediatric oncology and rare childhood cancers. For basic information see sympathetic nervous system resources, for treatment guidelines see clinical guideline sources, and for support networks see patient and family resources. Additional scientific and educational materials are available from specialty pages on imaging (diagnostic imaging), pathology (pathology references), and survivorship care (long-term follow-up).
Related articles
Author
AlegsaOnline.com Neuroblastoma: a pediatric cancer of the sympathetic nervous system Leandro Alegsa
URL: https://en.alegsaonline.com/art/69351
Sources
- emedicine.com : "eMedicine - Neuroblastoma : Article by Norman J Lacayo, MD"
- fordham.edu : "Cellular Communication: Unraveling the Secrets of Histone Proteins"
- linkinghub.elsevier.com : "MYCN-non-amplified metastatic neuroblastoma with good prognosis and spontaneous regression: a molecular portrait of stage 4S"
- doi.org : 10.1016/j.molonc.2008.07.002
- pubmed.ncbi.nlm.nih.gov : 19383347
- sciencedirect.com : "ScienceDirect - The Lancet : Neuroblastoma"
- emedicine.com : "eMedicine - Esthesioneuroblastoma : Article by Pavel Dulguerov, MD"