Joubert syndrome
Rare genetic neurodevelopmental disorder marked by cerebellar malformation (molar tooth sign), developmental delays, breathing and eye movement abnormalities, and variable multi-organ involvement.
Overview
Joubert syndrome is a rare inherited neurodevelopmental condition characterized by abnormal development of the midbrain and hindbrain. It is considered a ciliopathy, a group of disorders caused by defects in cellular cilia. First recognized in 1969, the syndrome remains uncommon: hundreds of affected individuals and families have been reported worldwide.
Image gallery
2 Images
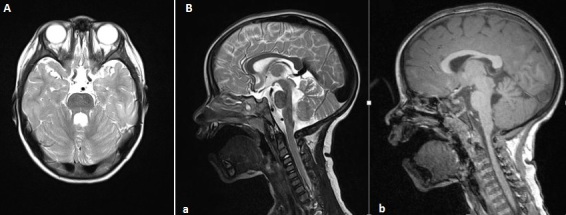
Key brain features
The hallmark radiologic sign is the so-called "molar tooth" appearance on axial magnetic resonance imaging, caused by underdevelopment of the cerebellar vermis and abnormal orientation of the brainstem. These structural differences alter coordination and motor control and help distinguish Joubert syndrome from other developmental disorders affecting the cerebellum.
Clinical presentation
Signs usually appear in infancy or early childhood and commonly include low muscle tone (hypotonia), delayed motor milestones, poor coordination (ataxia), and abnormal breathing patterns in neonates. Eye movement abnormalities such as oculomotor apraxia, as well as varying degrees of cognitive impairment, are frequent. Some people also develop retinal degeneration, kidney disorders, or liver fibrosis, making the syndrome a multi-system condition.
Genetics and cause
Joubert syndrome is genetically heterogeneous. Most cases follow an autosomal recessive pattern, and mutations at multiple genetic loci have been implicated—over twenty distinct loci have been associated with the disorder. These genes encode proteins important for the structure or function of primary cilia, which explains its classification as a genetic developmental ciliopathy.
Diagnosis and management
Diagnosis relies on clinical examination, characteristic neuroimaging (the molar tooth sign), and confirmatory molecular genetic testing when available. Management is supportive and multidisciplinary: therapies focus on respiratory support in early life, physical and occupational therapy for motor delays, vision monitoring, and surveillance for kidney or liver complications. There is currently no cure; treatment aims to maximize function and address organ-specific problems.
Prognosis and research
Outcomes vary widely depending on the severity of brain malformation and involvement of other organs. With early intervention and appropriate monitoring, many individuals can achieve improved motor skills and quality of life, though some face significant lifelong disabilities. Ongoing research into the underlying ciliary mechanisms and gene discovery aims to improve diagnosis, counseling, and future therapies. For more information and clinical resources see genetic resources and specialty centers listed at clinical registries and advocacy organizations (neurology references, ciliopathy networks).
Related articles
Author
AlegsaOnline.com Joubert syndrome Leandro Alegsa
URL: https://en.alegsaonline.com/art/51281
Sources
- arjournals.annualreviews.org : "The ciliopathies: an emerging class of human genetic disorders"
- doi.org : 10.1146/annurev.genom.7.080505.115610
- pubmed.ncbi.nlm.nih.gov : 16722803