Idiopathic pulmonary fibrosis: overview, features, diagnosis and management
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive scarring lung disease of unknown cause that primarily affects older adults; this article explains its features, diagnosis, treatment and prognosis.
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive disorder marked by scarring of the lung tissue that impairs breathing and gas exchange. The word pulmonary fibrosis describes the accumulation of fibrous tissue within the lung interstitium. In IPF the cause is unknown, which is why the adjective "idiopathic" is used. The condition most often develops in middle-aged to older adults and tends to follow a relentlessly progressive course with worsening exercise tolerance and breathlessness.
Image gallery
8 Images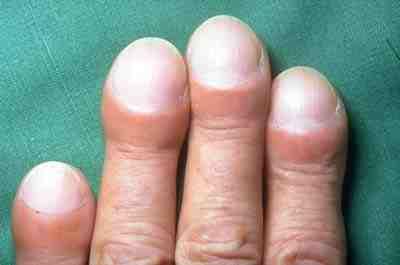





Clinical features and typical findings
Early symptoms are insidious. Patients commonly report:
- Progressive exertional dyspnea (shortness of breath)
- Persistent dry, nonproductive cough
- Fatigue and reduced exercise capacity
- On examination, fine inspiratory crackles (often described as "Velcro" crackles) and, in some cases, digital clubbing
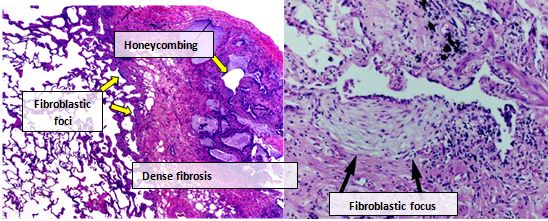
Pulmonary function tests typically show a restrictive pattern with reduced lung volumes and impaired gas transfer. High-resolution computed tomography (HRCT) often demonstrates a characteristic pattern called usual interstitial pneumonia (UIP), with reticulation, traction bronchiectasis and subpleural, basal predominance of fibrosis.
Diagnosis and differential considerations
Diagnosis of IPF requires the combination of compatible clinical, radiological and sometimes pathological findings together with exclusion of known causes of pulmonary fibrosis (such as connective tissue disease, drug toxicity or environmental exposures). HRCT has become central to diagnosis; when imaging is ambiguous, surgical lung biopsy or multidisciplinary assessment may be required. Distinguishing IPF from other interstitial lung diseases is important because prognosis and treatment differ.
Causes and pathophysiology
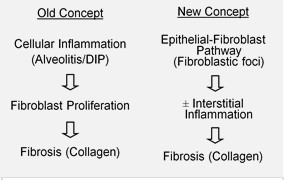
By definition, IPF lacks an identifiable external cause. Current understanding emphasizes repeated injury to the alveolar epithelium followed by aberrant repair, with activation of fibroblasts and excessive extracellular matrix deposition. Risk factors that increase the likelihood of disease include older age, male sex and a history of cigarette smoking. Genetic predisposition has been recognized in some familial cases, and research continues into molecular pathways that drive fibrosis.
Treatment, prognosis and supportive care
There is no cure for IPF in most patients, but several therapies can slow lung function decline. Two oral antifibrotic medications—nintedanib and pirfenidone—have been shown to reduce the rate of progression in many patients and are now widely used. Management also includes oxygen therapy for hypoxemia, vaccination, pulmonary rehabilitation, symptom control and assessment for lung transplantation in eligible candidates. Acute exacerbations can occur and represent a major cause of morbidity and mortality. Overall prognosis is variable but is often measured in years rather than decades; historically many patients have experienced a limited life expectancy after diagnosis, making early recognition and appropriate care important.
Importance, research and notable facts
IPF is one of the idiopathic interstitial pneumonias and forms part of the broader group of interstitial lung diseases (ILDs). It has attracted considerable research attention because of its poor outcomes and the need for better therapies. Clinical trials, biomarker discovery and efforts to refine diagnostic criteria continue. For patients and clinicians, key priorities are timely diagnosis, access to antifibrotic treatment where appropriate, comprehensive supportive care and consideration of lung transplantation when suitable.
For concise definitions and further reading about terms used here, see entries on pulmonary fibrosis, dyspnea and the implications of a poor prognosis.
Related articles
Author
AlegsaOnline.com Idiopathic pulmonary fibrosis: overview, features, diagnosis and management Leandro Alegsa
URL: https://en.alegsaonline.com/art/46533
Sources
- doi.org : 10.1164/rccm.2009-040GL
- pubmed.ncbi.nlm.nih.gov : 21471066
- ncbi.nlm.nih.gov : PubMed
- ncbi.nlm.nih.gov : "Idiopathic pulmonary fibrosis"
- doi.org : 10.1186/1750-1172-3-8
- pubmed.ncbi.nlm.nih.gov : 18366757
- doi.org : 10.1164/rccm.201506-1063ST