Chordoma: a rare malignant tumor of the spine and skull base
Chordoma is a rare, typically slow-growing malignant tumor arising from notochord remnants in the axial skeleton, often occurring at the skull base or sacrum and managed by surgery, specialized radiotherapy, and clinical trials.
Overview
Chordoma is an uncommon malignant tumor that arises from residual cells of the embryonic notochord. It characteristically appears along the axial skeleton, most often at the base of the skull (clivus), the sacrococcygeal region, or the mobile spine. Although it tends to grow slowly compared with many cancers, chordoma is locally aggressive, often recurs after treatment and can metastasize, particularly after long intervals.
Image gallery
5 Images



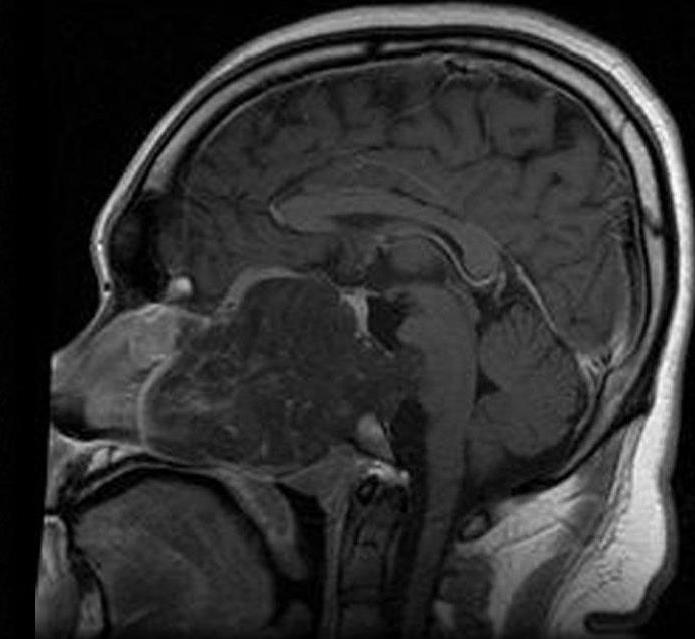
Anatomy and microscopic features
These tumors develop where notochordal tissue persisted during development. Microscopically chordoma cells are often vacuolated (so-called physaliphorous cells) and show both epithelial and mesenchymal markers. Immunohistochemistry for the transcription factor brachyury is widely used to support the diagnosis and to distinguish chordoma from other lesions.
Signs, symptoms and diagnosis
Clinical features depend on location. Typical presentations include:
- Skull base (clival): headache, double vision or other cranial nerve deficits, difficulty swallowing.
- Spine: localized pain, radicular symptoms or myelopathy.
- Sacrum: low back pain, bowel or bladder dysfunction, and tailbone pain.
Diagnosis relies on imaging (MRI and CT to define extent and bone involvement) followed by biopsy with histology and immunostaining. The combination of imaging features and brachyury positivity helps to separate chordoma from chondrosarcoma and metastatic tumors.
Treatment options
Management is individualized and often multimodal. Key approaches include:
- Maximal safe surgical resection, aiming for wide or en bloc removal when feasible.
- High‑precision radiotherapy—such as proton beam or carbon ion therapy—to control residual disease or treat unresectable tumors.
- Systemic therapy is limited; targeted agents and clinical trials are options for advanced or metastatic disease.
Because complete resection can be difficult near critical structures (skull base, sacrum), long-term follow-up with periodic imaging is essential.
Prognosis, research and notable facts
Prognosis varies with tumor size, location, and completeness of resection. Long-term survival is possible, but local recurrence is common and late metastases can occur. Research focuses on the molecular drivers of chordoma—particularly the role of the brachyury (T) gene, which has been implicated in tumor biology and, in some familial cases, shown to be duplicated—plus development of targeted therapies and advanced particle radiotherapy techniques.
Because chordoma is rare and complex to treat, care is often best delivered in specialized centers and through multidisciplinary teams; for general patient information and resources see further reading and support.
Tags
Related articles
Author
AlegsaOnline.com Chordoma: a rare malignant tumor of the spine and skull base Leandro Alegsa
URL: https://en.alegsaonline.com/art/19978