Sickle-cell disease
Inherited blood disorder in which abnormal hemoglobin causes red blood cells to deform, producing chronic anemia, painful crises, organ damage, and increased infection risk; varied severity and treatments exist.
Overview
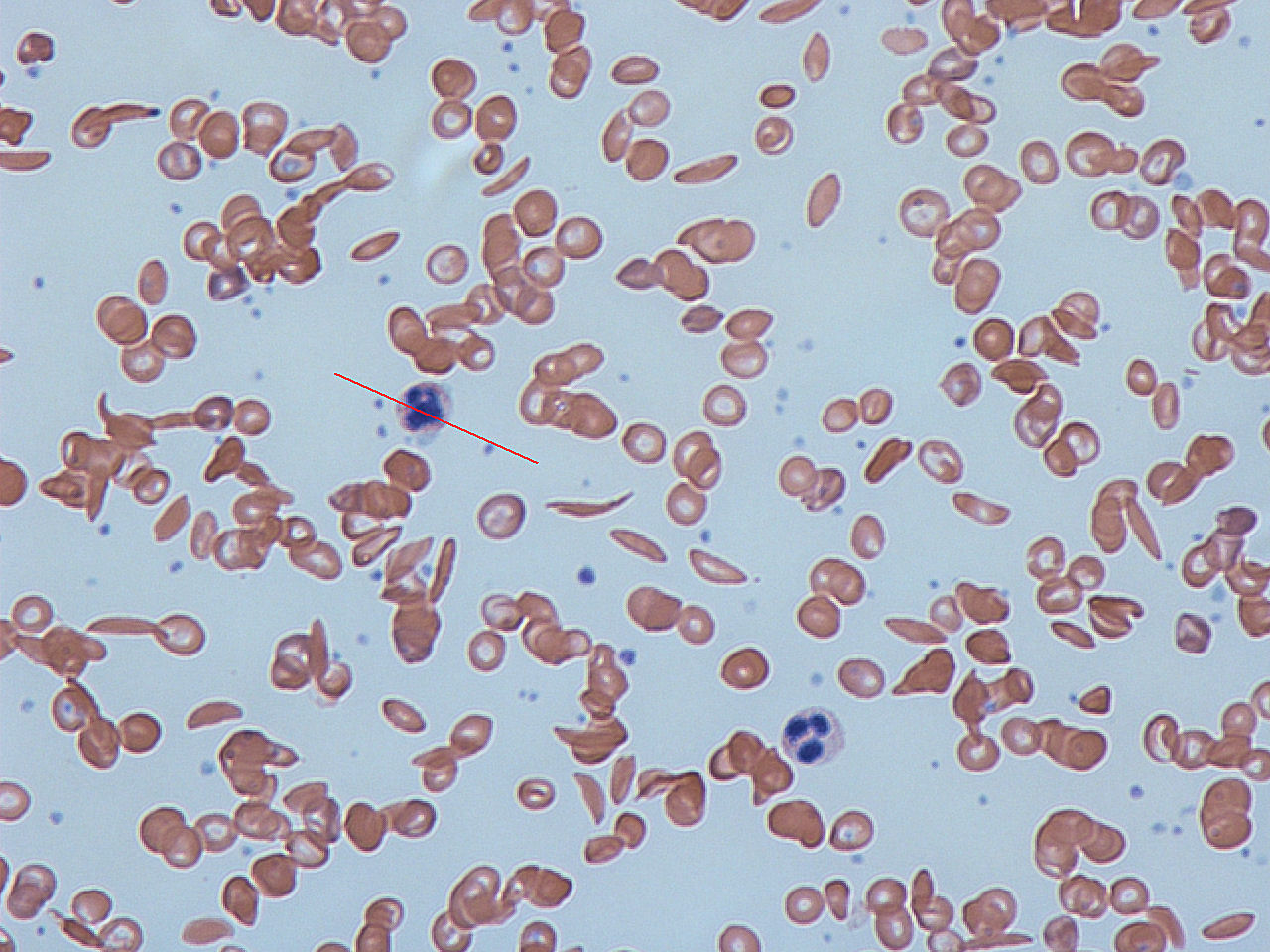
Sickle-cell disease is an inherited disorder of hemoglobin that causes red blood cells to adopt a rigid, crescent or "sickle" shape under low-oxygen conditions. These altered cells are less flexible and more fragile than normal biconcave erythrocytes, which leads to chronic hemolytic anemia and periodic blockages in small blood vessels (vaso-occlusion). Recurrent vaso-occlusion and hemolysis produce pain, tissue ischemia, and progressive organ damage over a person's lifetime.
Image gallery
8 Images





Causes and genetics
The condition results from a mutation in the beta-globin gene that gives rise to hemoglobin S (HbS). Sickle-cell disease most commonly occurs when an individual inherits two copies of the mutated gene (homozygous HbSS). People with one normal and one mutated gene (sickle-cell trait, HbAS) usually have few symptoms but can transmit the mutation to offspring. The trait is common in regions that historically had high malaria rates because carriers have some protection against severe malaria.
Clinical features and complications
Typical manifestations include chronic anemia, acute painful episodes called vaso-occlusive crises, and increased susceptibility to infection—especially early in life when the spleen is affected. Over time the disease can cause strokes, chronic kidney disease, pulmonary hypertension, leg ulcers, avascular necrosis of bones, and progressive organ dysfunction. Severity varies widely between individuals and can be influenced by coexisting genetic and environmental factors.
Diagnosis and screening
Diagnosis is established by examining a blood smear and confirming abnormal hemoglobin with tests such as hemoglobin electrophoresis or high-performance liquid chromatography. Newborn screening programs in many countries identify affected infants early so that preventive measures can begin. For an introduction to normal red blood cell form and function see red blood cells; for descriptions of cell shape and deformation see cell morphology.
Treatment and prevention
Management combines symptom control, prevention of complications, and disease-modifying therapies. Common approaches include:
- Hydroxyurea, which raises fetal hemoglobin and reduces crises;
- Regular or episodic blood transfusions to prevent or treat severe anemia and reduce stroke risk;
- Preventive antibiotics and vaccinations to lower infection risk in young children;
- Pain management, hydration, and treatment of acute complications;
- Curative options such as hematopoietic stem cell transplantation in selected patients and emerging gene therapies under investigation.
For authoritative guidance on care and current therapeutic advances consult clinical resources and specialist centers: treatment and resources.
History and public health
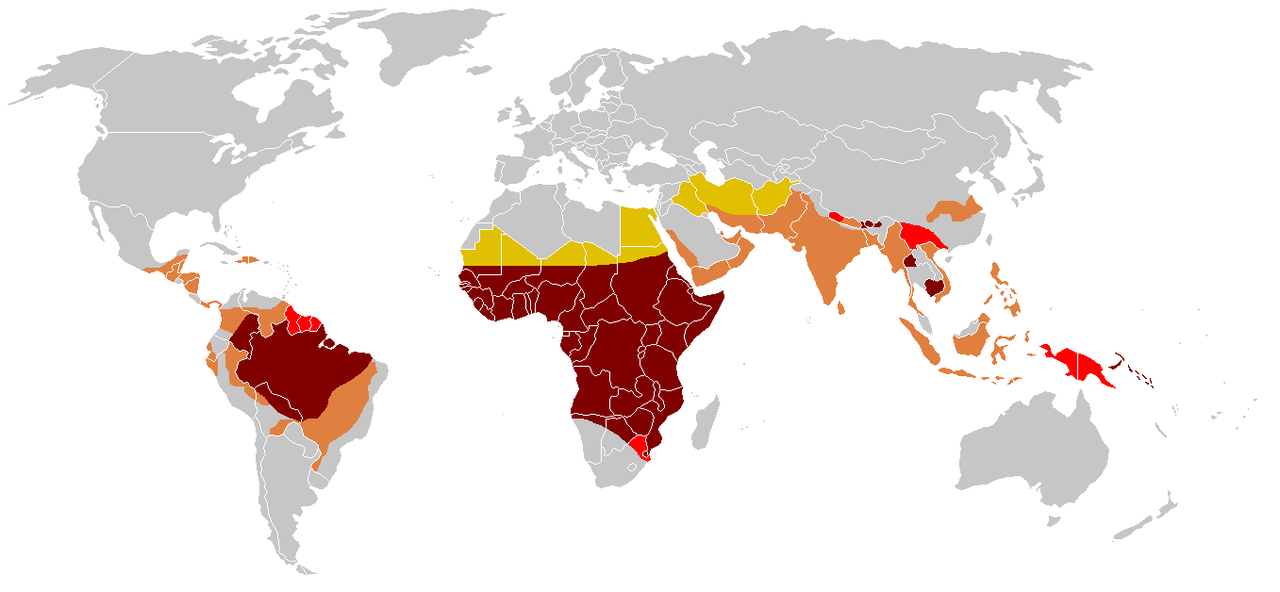
Recognition of the disease dates back more than a century; subsequent research established its molecular basis and inheritance pattern. Today it is an important global public‑health issue, particularly in sub-Saharan Africa, parts of the Mediterranean, the Middle East, and India. Public-health strategies emphasize newborn screening, vaccination, access to preventive care, and genetic counseling to reduce morbidity and improve life expectancy.
Notable distinctions
"Sickle-cell disease" is an umbrella term that includes several genotypes (for example HbSS, HbSC, HbSβ-thalassemia) with overlapping but distinct clinical courses. Differentiating sickle-cell disease from sickle-cell trait is important for prognosis, counseling, and family planning.
Questions and answers
Q: What is sickle-cell disease?
A: Sickle-cell disease is a genetic disease that affects red blood cells, changing them from flexible disks into rigid crescents.
Q: How does sickle-cell disease affect the body?
A: When many red blood cells take on the sickle shape, veins can become blocked, causing damage to many organs. The damage increases over time and can lead to an early death.
Q: What causes sickle-cell disease?
A: Sickle-cell disease is caused by a genetic mutation that affects the production of hemoglobin, a protein found in red blood cells.
Q: Can sickle-cell disease be cured?
A: There is currently no cure for sickle-cell disease, but there are treatments available to help manage the symptoms and complications of the disease.
Q: How is sickle-cell disease diagnosed?
A: Sickle-cell disease is typically diagnosed through a blood test that measures the amount of abnormal hemoglobin in the blood.
Q: Is sickle-cell disease contagious?
A: No, sickle-cell disease is not contagious. It is an inherited condition that is passed down from parents to their children.
Q: Can sickle-cell disease be prevented?
A: While sickle-cell disease cannot be prevented, prenatal screening and genetic counseling can help parents make informed decisions about having children and managing the disease.
Related articles
Author
AlegsaOnline.com Sickle-cell disease Leandro Alegsa
URL: https://en.alegsaonline.com/art/90200
Sources
- nejm.org : nejm.org/doi/full/10.1056/NEJM200006223422502
- jci.org : "The impact of malaria parasitism: from corpuscles to communities"
- doi.org : 10.1172/JCI38307
- ncbi.nlm.nih.gov : 2735907
- pubmed.ncbi.nlm.nih.gov : 19729847
- content.nejm.org : "Mortality in sickle cell disease. Life expectancy and risk factors for early death"
- doi.org : 10.1056/NEJM199406093302303
- worldcat.org : 0028-4793
- pubmed.ncbi.nlm.nih.gov : 7993409