Prion: proteinaceous infectious agents that cause neurodegenerative disease
Prions are misfolded, infectious proteins that induce abnormal folding of normal proteins, causing fatal neurodegenerative conditions in mammals and other organisms; they are distinct from viruses and bacteria.
A prion is an infectious protein particle whose pathogenicity comes from an abnormal three‑dimensional shape rather than from nucleic acid. The term is short for "proteinaceous infectious particle" and emphasizes that the infectious entity is a misfolded protein rather than a conventional microbe such as a virus or bacterium. The concept of an infectious protein helped explain unusual, transmissible diseases that attack the brain and nervous system. For a general reference to the biochemical nature of prions see infectious protein.
Image gallery
6 Images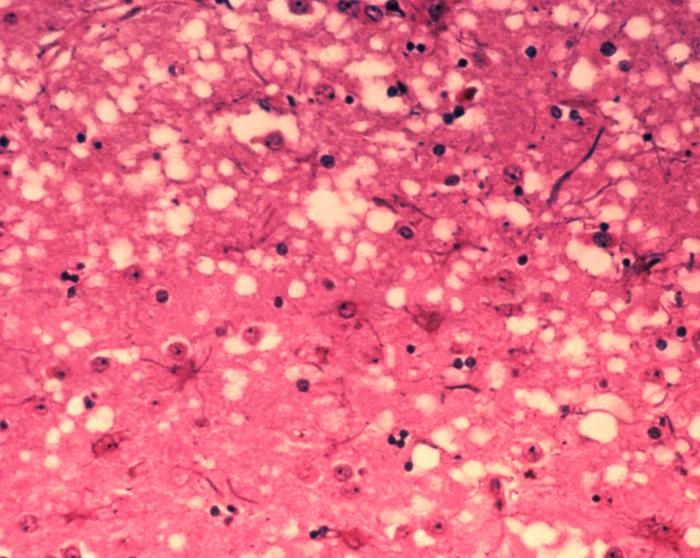



Characteristics and mechanism
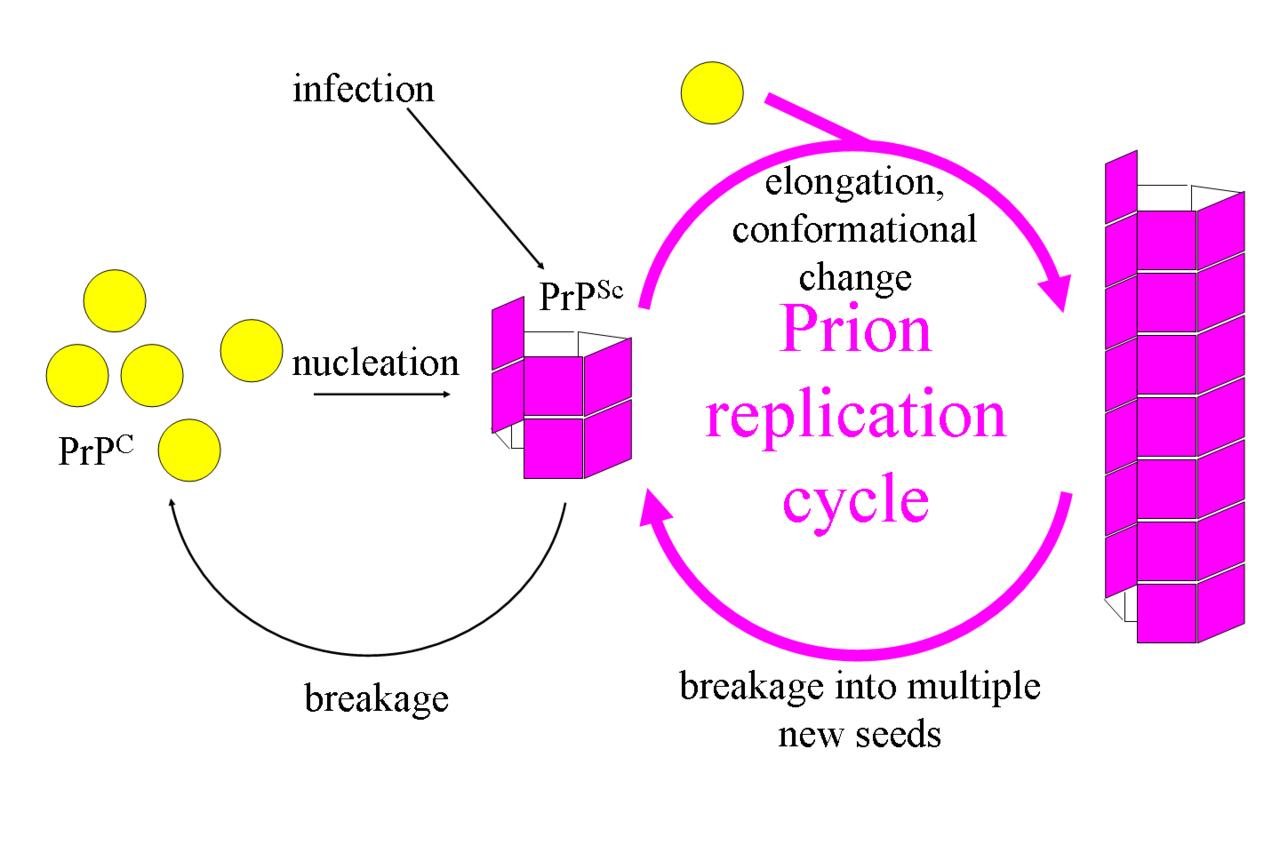
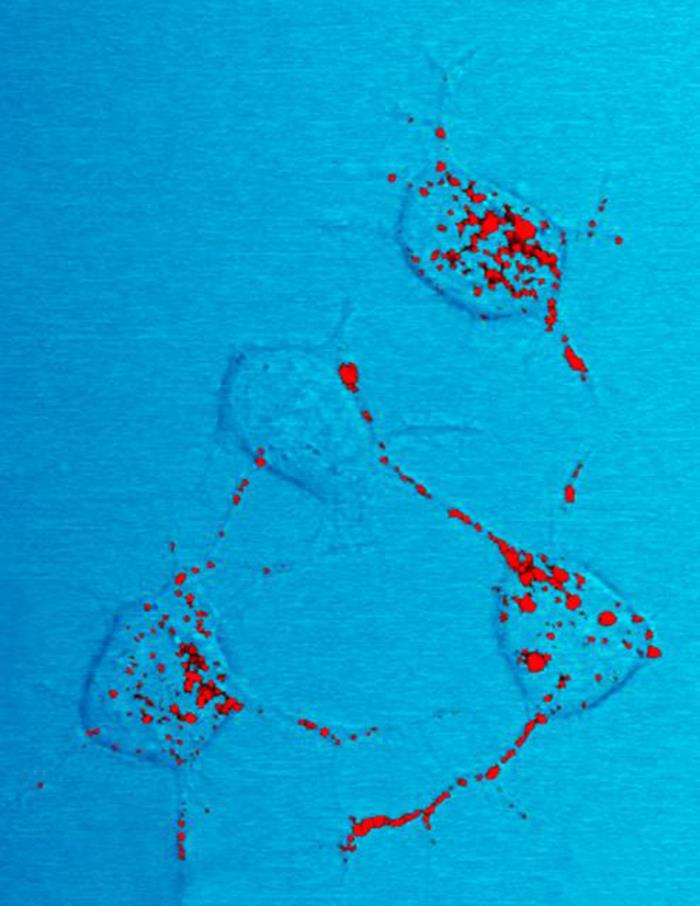
Prions arise when a normally folded host protein adopts a self‑propagating, misfolded conformation. The normal cellular form, often called PrPC in mammals, is rich in alpha helical structure; the disease‑associated form is enriched in beta sheet and tends to form insoluble aggregates. This change in secondary structure — from alpha helices to beta sheets — underlies the ability of the abnormal protein to convert the normal form into additional copies of the misfolded state.
- They lack nucleic acids and reproduce by templated conformational change rather than by gene replication.
- Misfolded prion proteins are often resistant to heat, many proteases and common disinfectants, which complicates decontamination.
- Prions primarily damage the brain and other neural tissues, producing characteristic sponge‑like (spongiform) lesions.
Diseases and examples
In mammals, prion infections cause a group of conditions known as transmissible spongiform encephalopathies. These include classical farm and wildlife diseases such as scrapie in sheep, bovine spongiform encephalopathy (BSE or "mad cow" disease) in cattle, and chronic wasting disease in deer and elk. In humans they are associated with Creutzfeldt‑Jakob disease and related syndromes, and historically with kuru observed in certain populations. Collectively these are sometimes referred to as spongiform encephalopathies.
History, discovery and research
The prion hypothesis was proposed to explain infectious neurodegeneration that could not be accounted for by conventional pathogens. The idea provoked controversy but gradually gained evidence through biochemical and genetic studies. Intensive work in molecular biology and biophysics has clarified many aspects of prion structure and propagation, while other research has shown that protein‑based inheritance occurs in simpler organisms like yeast, where prion‑like states can alter cell physiology without causing disease.
Transmission, diagnosis and public health
Transmission routes vary: ingestion of contaminated tissue, iatrogenic exposure (medical instruments or transplanted tissues), and inheritance via mutations in the gene encoding the prion protein are documented. Prion diseases typically have long incubation periods, progressive neurological decline and are fatal. Diagnostic approaches rely on clinical assessment, neuroimaging, laboratory assays that detect abnormal prion seeds or surrogate markers, and definitive diagnosis by neuropathology. Because no curative therapy exists, control emphasizes surveillance, safe handling of potentially infectious materials and measures to prevent cross‑transmission.
Notable distinctions and ongoing questions
Prions are notable for being infectious without nucleic acid, for their unusual resistance to routine sterilization, and for demonstrating that information can be encoded in protein conformation. Active areas of research include the precise molecular steps of templated misfolding, factors that modulate species barriers to transmission, and strategies to detect and neutralize prion seeds. For broader overviews and resources see basic descriptions and specialized literature linked at neuropathology sources or molecular reviews accessed via neural biology collections.
Further reading and surveillance updates are available from scientific reviews and public health bodies; examples of focused discussions and summaries can be found by following portals indexed at spongiform disease summaries, research repositories on animal prion diseases, historical case studies such as kuru, and technical pages about protein folding and structure secondary structure. For current experimental techniques and deeper mechanistic studies consult pages on molecular biology methods.
The simplified prion hypothesis and features of prion diseases.
One of the numerous proteins found in the animal body is called PrPC (prion protein cellular). It is mainly found in the nervous system, especially in the brain. Prions differ more or less slightly between different animal species and possibly also within an animal species. PrPC is mainly found on the cell surface and protects the cells from divalent copper ions, H2O2 and free radicals. Furthermore, it is thought to be one of the first sensors in the cellular defense against reactive oxygen and free radicals and has effects on the enzymatic degradation of free radicals.
If this normal protein PrPC comes into contact with a protein called PrPSc (Prion Protein Scrapie; pathogenic form of the prion protein, which was first found in the form in animals suffering from scrapie), PrPC takes on the form of PrPSc, "it flips over", it changes its conformation. A chain reaction develops in which more and more PrPC is converted to PrPSc. Large amounts of PrPSc are destructive to the brain because they are insoluble and are deposited in cells. As a result, these cells die; holes appear in the brain, a sponge-like structure develops. Hence the name of this disease: spongiform encephalopathy, spongy brain disease. Prion diseases are always fatal.
The initiation of the disease can be done in three ways, which is unique among all diseases:
- sporadic, i.e. random or without an identifiable cause: PrPC folds over into PrPSc "by chance" and thereby triggers the chain reaction. One example is the classic form of Creutzfeldt-Jakob disease (sCJD).
- genetic, i.e. due to an "error" in the genetic material: the PRNP gene, on which the information for the production of PrPC is deposited in the form of DNA, may contain a mutation. The then altered protein is more susceptible to conversion into PrPSc. The mutation can be inherited from parents to children. Examples are the familial form of Creutzfeldt-Jakob disease (fCJD), Gerstmann-Sträussler-Scheinker syndrome (GSS) and fatal familial insomnia (FFI).
- by transmission or "infection": if PrPSc is introduced from outside, this can in turn convert one's own PrPC into PrPSc, although not under all circumstances. It depends on how much PrPSc is ingested, how it is ingested and exactly what type of PrPSc is ingested. Infection in everyday contact with patients is not possible. However, transmission is quite possible if material with a high PrPSc content, e.g. the brain of sick animals or humans, enters the bloodstream or, in the worst case, directly into the brain. This is the case, for example, with the iatrogenic form of Creutzfeldt-Jakob disease (iCJD). During brain surgery, inadequately sterilized instruments have inadvertently introduced prions from diseased individuals into the brains of healthy individuals. Another example is Kuru, a disease in Papua New Guinea; members of the affected tribe introduced the brains of deceased persons and thus high amounts of PrPSc to themselves as part of cultural rites. The most prominent example, however, is certainly BSE ("mad cow disease"). According to the theory that is widely accepted in science, these PrPSc were spread by meat-and-bone meal, which was produced, among other things, from the carcasses of sheep suffering from scrapie and then fed to cattle. In addition, in the United Kingdom, the procedures for the production of meat-and-bone meal had been modified (lower temperature or pressure) in such a way that these PrPSc were able to survive the production process. To a lesser extent, other animals (cats, zoo animals) have probably been infected by this meat-and-bone meal or by feeding parts of diseased cattle. Finally, in Great Britain in particular, people have probably contracted a new form of Creutzfeldt-Jakob disease, the "new variant" (nvCJD or vCJD), as a result of eating meat or brain or spinal cord tissue from BSE cows. In experiments with mice, Swiss researchers discovered a new route of infection, via the air we breathe. Whether such an infection is also possible in humans is still controversial.
The familial forms of prion diseases can also be transferred in experiments. For example, PrPSc, which develops in a human due to genetic disposition, can trigger the disease in mice if it has previously been injected into the brain.
Prions are very resistant to standard disinfection or sterilisation procedures, which was also a reason for the iCJD cases and the BSE crisis. Today, strict regulations are in place for the sterilisation of material that has come into contact with tissue that may contain prions, in line with the difficulty of inactivating prions. Since infections are often only discovered during post-mortem examinations, corpses must always be handled as if an infection were present. Due to the high resistance of the pathogens, the strictest measures apply to the handling of cadavers with confirmed prion diseases as well as to contact with potentially infectious tissue, up to and including excision of the affected area.
Today, the prion hypothesis is considered relatively certain. However, the possibility that another factor besides the PrPSc plays a role cannot yet be definitively ruled out. After even the intensive search for viruses, viroids or nucleic acid was unsuccessful in the first place, there are hardly any scientists who continue to pursue this path. Outsider opinions occasionally circulate among the public, such as the organophosphate theory, according to which BSE is linked to insect venoms, for which, however, there is no scientific evidence.
History of prion research
Individual prion diseases were described a long time ago (scrapie, the prion disease of sheep, in 1759 by Johann George Leopoldt; CJD in 1920 by Hans Gerhard Creutzfeldt), without anything being known about the cause of these diseases or being able to classify them into a group. After the transmissibility of scrapie was demonstrated in 1932 and kuru was first described in 1957, the similarity of these diseases was noted by William J. Hadlow in the late 1950s and kuru was also transferred experimentally to monkeys. In 1966, Tikvah Alper and coworkers determined that the pathogen was too small to be a virus and apparently contained no nucleic acid. Therefore, a "protein only" hypothesis for the pathogen emerged, although how such a protein could replicate remained unclear. Many therefore assumed lentiviruses to be the most likely cause.
The "prion hypothesis" published in 1982 by Stanley Prusiner, who followed up on the work of Daniel C. Gajdusek, who had already unknowingly discovered a pathogenic prion, was initially received critically by the scientific community, as a nucleic acid-free infectious agent had not been conceivable until then. In retrospect, however, this hypothesis proved to be groundbreaking, and in 1997 Prusiner was awarded the Nobel Prize for his work in the field of prion research. In the years following the establishment of this hypothesis, evidence for its accuracy was obtained in numerous experiments, although no definitive proof was obtained. In 1986 the BSE epidemic began in Great Britain, and in 1996 the first cases of vCJD were described.
The proof that recombinant prion protein can cause disease (thus fulfilling Koch's postulate) was achieved in 2010.
Politicians tried to make up for their failures in prevention and consumer protection, among other things, by spending generously in the field of prion research. Numerous working groups were newly established, centres built and alliances founded. Prion research was intensified and accelerated.
In 2007, doubts arose as to whether the pathogenic prion content of a tissue correlates with its infectivity in every case.
New research results around the US-American researcher Susan Lindquist show that prions play an important role in neurogenesis (development of new nerve cells in the brain).
Related articles
Author
AlegsaOnline.com Prion: proteinaceous infectious agents that cause neurodegenerative disease Leandro Alegsa
URL: https://en.alegsaonline.com/art/79248
Sources
- ncbi.nlm.nih.gov : "Prions"
- ui.adsabs.harvard.edu : 1998PNAS...9513363P
- doi.org : 10.1073/pnas.95.23.13363
- pubmed.ncbi.nlm.nih.gov : 9811807