Acromegaly — causes, symptoms, diagnosis, and treatment
Acromegaly is a rare hormonal disorder caused by excess growth hormone after puberty, producing characteristic tissue overgrowth, metabolic effects, and increased health risks if untreated.
Acromegaly is an endocrine disorder that results from chronic excess production of growth hormone (GH) by the anterior pituitary gland after the normal completion of skeletal growth. Persistent GH overproduction stimulates the liver to make insulin-like growth factor 1 (IGF-1), which mediates many of the growth and metabolic effects. When excess GH occurs before the growth plates close in childhood or adolescence the condition produces generalized tall stature known as gigantism; after puberty the same hormonal imbalance causes the characteristic pattern of bone and soft-tissue enlargement called acromegaly.
Image gallery
8 Images






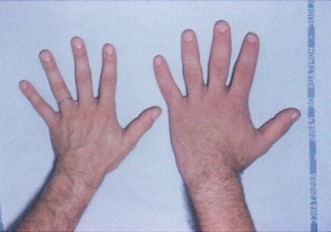
Causes and mechanism
The most frequent cause of acromegaly is a benign GH-secreting tumor of the pituitary gland, known as a pituitary adenoma. Less commonly, GH excess can arise from rare tumors outside the pituitary or from hypothalamic pathology that stimulates GH release. The sustained elevation of GH and IGF-1 leads to progressive enlargement of skin, connective tissue and certain bones, along with metabolic disturbances such as insulin resistance.
Signs and symptoms
Changes usually appear slowly and may take years to be recognized. Typical features include enlargement of the hands and feet, widening of the jaw and spacing of the teeth, pronounced brow and nose, thickened skin, and increased soft-tissue volume of the tongue and lips. Patients commonly report joint pain, excessive sweating, fatigue, headache and changes in shoe or ring size. Systemic complications include sleep apnea, carpal tunnel syndrome, hypertension, cardiomyopathy, and impaired glucose tolerance or diabetes.
Diagnosis
Diagnosis relies on a combination of clinical assessment and biochemical testing. Measurement of serum IGF-1 is a sensitive screening test because IGF-1 levels remain elevated throughout the day. If IGF-1 is high or suspicion remains, dynamic testing with an oral glucose tolerance test is used to demonstrate the failure of GH to suppress. Imaging, most often magnetic resonance imaging (MRI) of the pituitary, identifies and characterizes an adenoma when present. Because the disorder evolves slowly, diagnosis is frequently delayed for many years.
Treatment and prognosis
Therapy aims to control GH/IGF-1 excess, reduce tumor mass when present, relieve symptoms and prevent or manage complications. First-line treatment for many patients is transsphenoidal surgical removal of a pituitary adenoma. Medical therapies include somatostatin analogs (which reduce GH secretion), GH receptor antagonists (which block GH action), and dopamine agonists in selected cases. Radiotherapy is an option when surgery and medication do not achieve control. With modern multidisciplinary care, many patients experience symptom improvement and reduced disease-related mortality, but long-term follow-up is necessary to monitor for recurrence and manage comorbid conditions.
History, prevalence and notable facts
Acromegaly is uncommon and is most often detected in middle-aged adults. Historical descriptions of the facial and physical changes date back centuries, but effective biochemical and surgical treatments developed during the 20th century greatly improved outcomes. Key distinctions—most importantly the difference between acromegaly and childhood gigantism—are based on whether excess GH occurs before or after the closure of growth plates. Early recognition improves the chances of restoring hormone balance and preventing long-term complications.
- Key points: caused mainly by pituitary adenoma; diagnosed by elevated IGF-1 and GH suppression testing; treated with surgery, medication, and sometimes radiotherapy.
Questions and answers
Q: What is acromegaly?
A: Acromegaly is a medical condition that occurs when the pituitary gland makes too much growth hormone after puberty.
Q: What causes gigantism?
A: Gigantism is caused by an excessive amount of growth hormone produced by the pituitary gland before puberty.
Q: What is the most common cause of acromegaly?
A: The most common cause of acromegaly is the presence of a pituitary adenoma, a tumor on the pituitary gland.
Q: Who is most at risk of developing acromegaly?
A: Acromegaly is most commonly diagnosed in middle-aged adults.
Q: What are the potential consequences of untreated acromegaly?
A: Untreated acromegaly can result in severe disfigurement, serious complications, and early death.
Q: Why is acromegaly difficult to diagnose?
A: Acromegaly is hard to diagnose when it is just beginning and is usually not found until 10-12 years after it starts.
Q: What are the visible symptoms of acromegaly?
A: The most noticeable symptom of acromegaly is changes in a person's physical appearance, particularly in their face.
Related articles
Author
AlegsaOnline.com Acromegaly — causes, symptoms, diagnosis, and treatment Leandro Alegsa
URL: https://en.alegsaonline.com/art/762