Amyloidosis: causes, types, diagnosis, and clinical significance
Amyloidosis is a group of disorders caused by abnormal protein fibril deposits in tissues. This article explains types, pathology, symptoms, diagnosis, treatment approaches and notable facts.
Amyloidosis describes a set of conditions in which normally soluble proteins misfold and assemble into insoluble fibrils that deposit in tissues. These deposits — called amyloid — disrupt normal organ structure and function. Amyloidosis can be localized to a single tissue or organ (local amyloidosis) or involve multiple organs throughout the body (systemic amyloidosis). The disorder is uncommon, has many forms, and produces a wide spectrum of signs and severity, so recognition and diagnosis can be challenging.
Image gallery
10 Images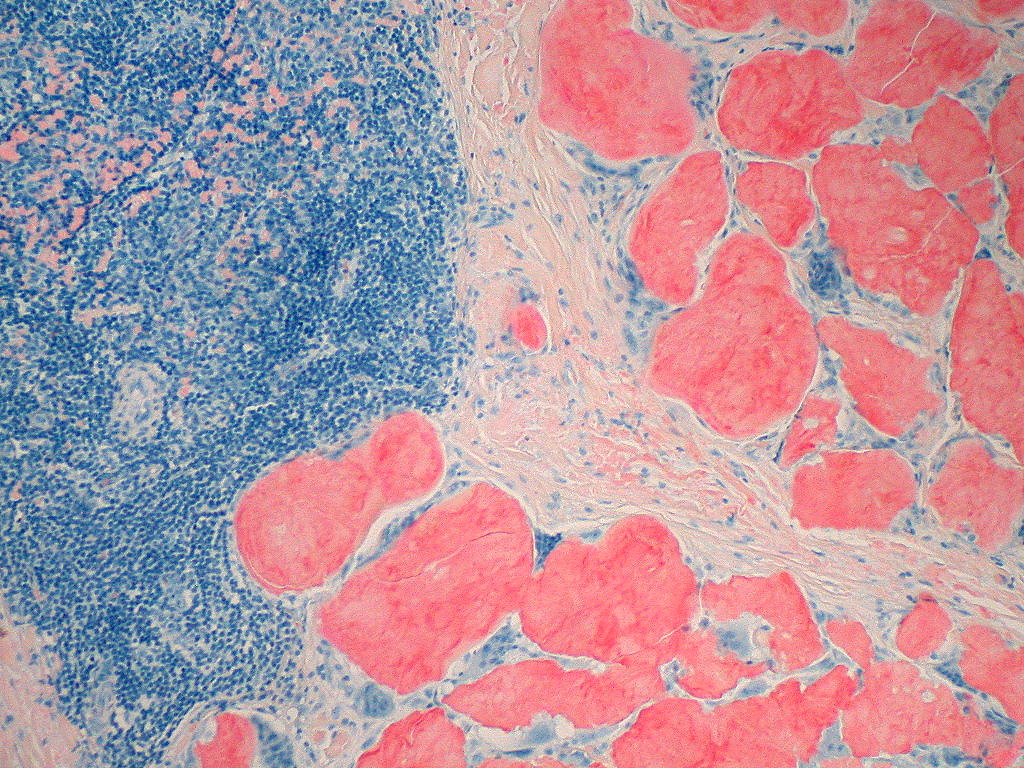





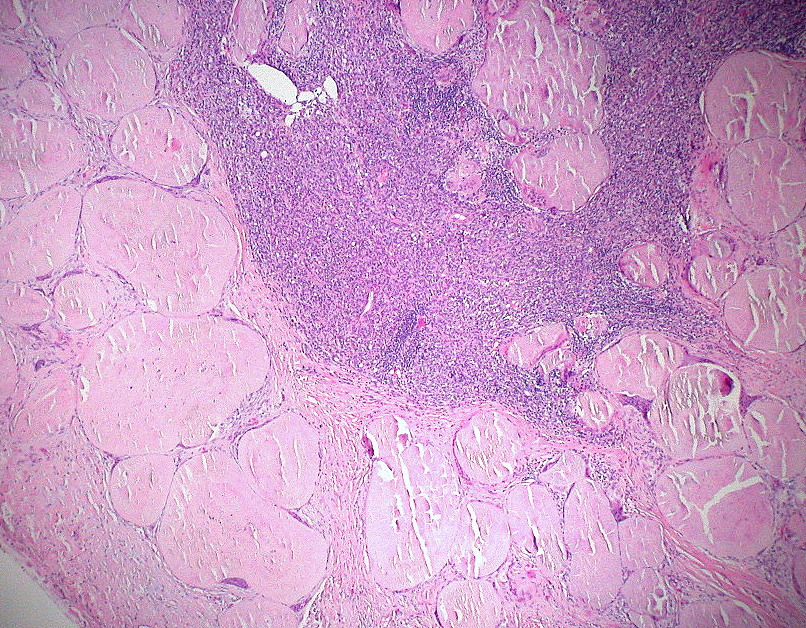
Characteristics and pathology
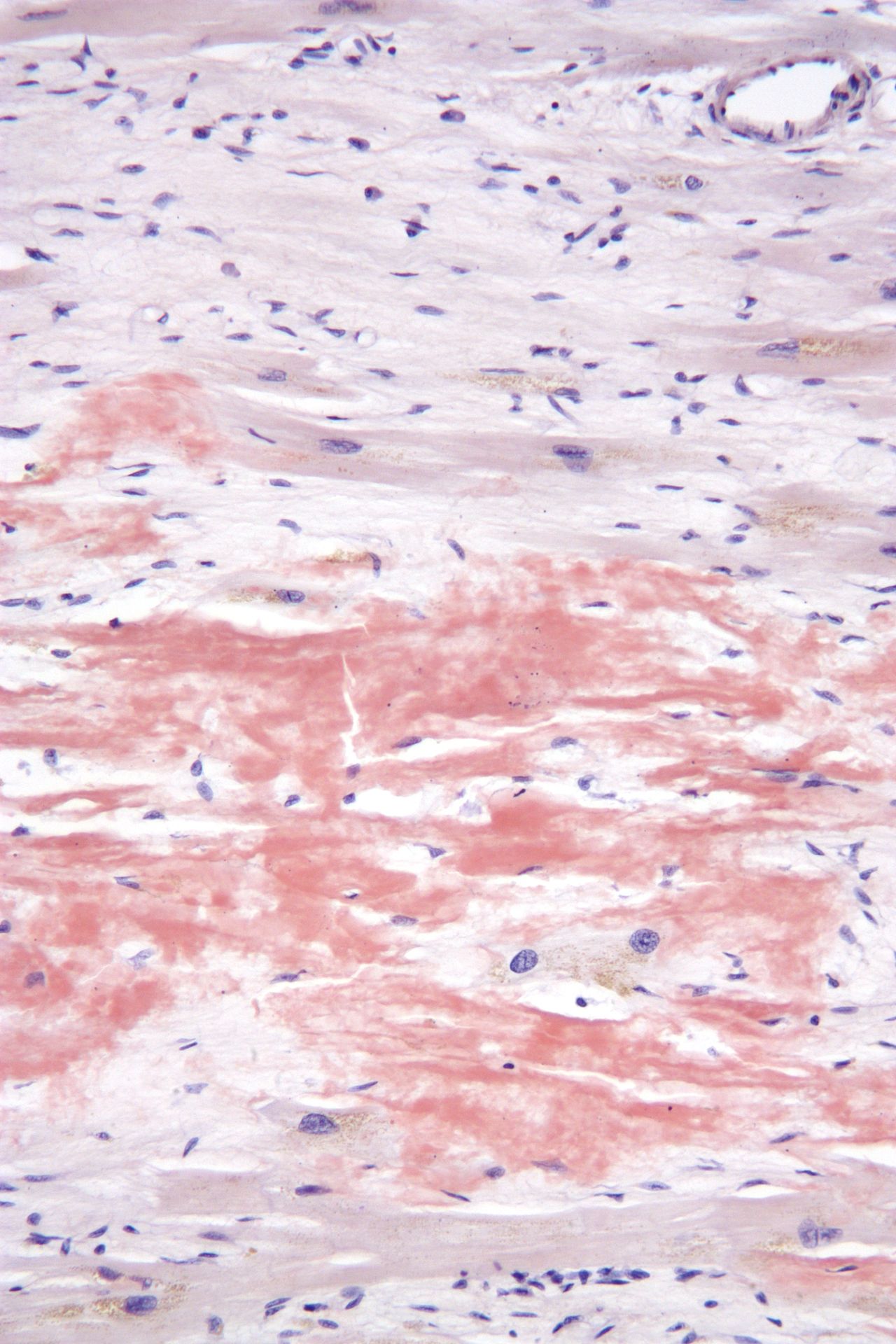
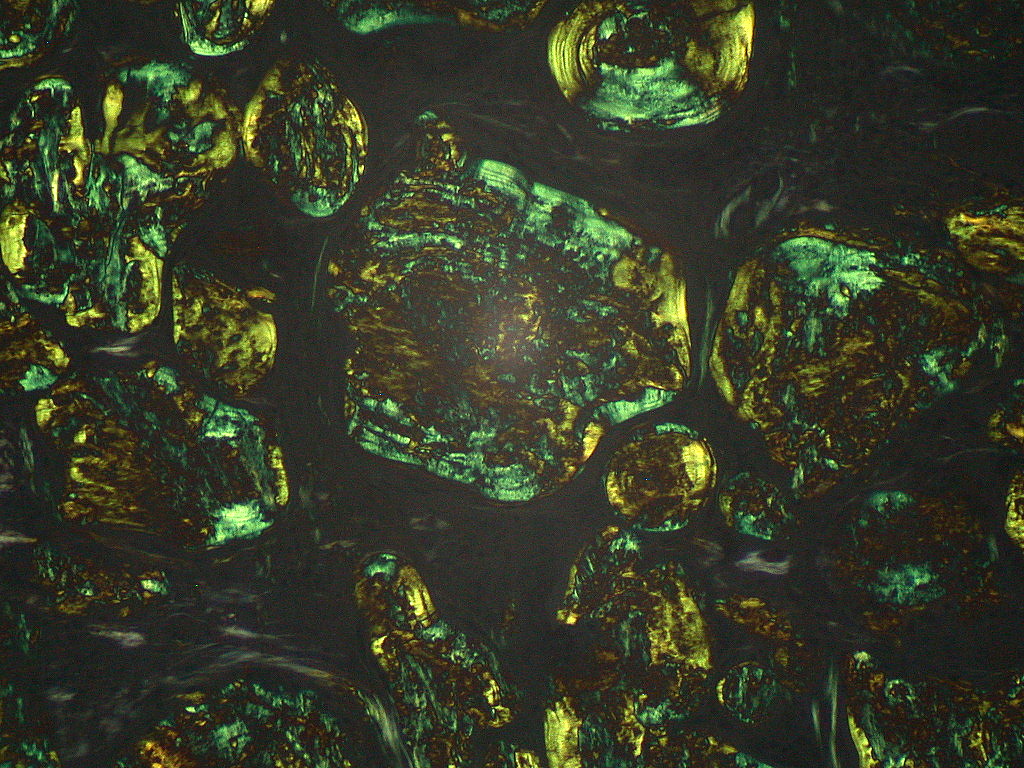
At the microscopic level amyloid fibrils have a characteristic beta‑sheet conformation and bind certain stains that allow pathologists to identify them in biopsy specimens. When examined under polarized light after Congo red staining, amyloid classically shows an "apple‑green" birefringence. The deposits are extracellular and may replace normal tissue, impairing organ function. Organs commonly affected in systemic disease include the kidneys, heart, liver, peripheral nerves and gastrointestinal tract; localized deposits may occur in skin, the respiratory tract, or other single sites.
Classification and common types
Amyloidosis is classified according to the biochemical identity of the fibril protein. More than two dozen human proteins are known to form amyloid, and the most frequently encountered types include:
- AL (light‑chain) amyloidosis — fibrils derived from immunoglobulin light chains produced by clonal plasma cells; often systemic and may involve heart and kidneys.
- AA (secondary) amyloidosis — fibrils composed of fragments of serum amyloid A protein, typically associated with chronic inflammatory conditions.
- ATTR amyloidosis — fibrils derived from transthyretin, which can be the normal (wild‑type) protein or a variant from a genetic mutation; often affects the heart and nerves.
- Localized amyloidoses — produced by local precursor proteins at the site of deposition, such as amyloid in the brain associated with Alzheimer-type pathology or in localized nodules.
Because the underlying proteins and mechanisms differ, accurate typing of amyloid is essential for prognosis and therapy.
Clinical presentation and diagnosis
Symptoms depend on which organs are affected and the extent of deposition. Renal involvement typically presents with proteinuria and progressive kidney dysfunction; cardiac involvement can cause restrictive cardiomyopathy and arrhythmias; neuropathy often produces numbness, tingling and autonomic symptoms. Because early manifestations are nonspecific, diagnosis often requires a combination of clinical suspicion, imaging, laboratory studies, and tissue biopsy. Fat pad aspiration or affected‑organ biopsy with Congo red staining and additional biochemical or immunohistochemical methods is used to confirm and type the amyloid protein.
Treatment and prognosis
Treatment strategies aim to reduce production of the offending precursor protein and support affected organs. For AL amyloidosis, therapies that suppress the abnormal plasma cell clone (for example, chemotherapy regimens used in plasma cell disorders) can reduce new light‑chain production. For ATTR amyloidosis, options include medicines that stabilize transthyretin, agents that reduce its production, or, in selected patients, liver transplantation or cardiac interventions. Management also addresses organ‑specific complications such as heart failure or renal impairment. Prognosis varies greatly by type, organ involvement and response to therapy; early diagnosis and targeted treatment improve outcomes.
History, epidemiology and notable facts
The word "amyloid" was introduced in the mid‑19th century to describe starch‑like deposits seen in tissues. Modern understanding identifies amyloid as proteinaceous fibrils with distinctive structural properties. Amyloidosis is rare compared with many chronic diseases, and its true prevalence depends on the subtype and population studied. Awareness of the diversity of amyloid‑forming proteins has increased with advances in biochemical and genetic testing.
Several public figures have been associated with amyloidosis in media reports; examples include Martin McGuinness, David Lange, Robert Jordan and Robert P. Casey. Their cases have helped raise public awareness of the disease.
Because amyloidosis spans multiple medical specialties and has many subtypes, effective care often requires coordination between hematologists, cardiologists, nephrologists, neurologists and pathologists. Ongoing research into the molecular mechanisms of protein misfolding is driving development of new diagnostics and therapies aimed at halting fibril formation or clearing deposits.


Cause and origin
Amyloidosis is caused by a disorder in the folding of a normally soluble protein. Several different diseases can trigger the disease through overproduction, lack of/reduced degradation or impaired excretion of certain proteins. These proteins are usually present in the blood plasma in a dissolved form. However, if their concentration increases, they also reach the surrounding tissue, where enzymes attack them. The aggregation of the resulting amino acid chains in the area of the β-sheet structures results in the formation of insoluble complexes in the form of microscopic fibres (fibrils).
Since the fibrils are resistant to the defense mechanisms (phagocytosis and proteolysis), they can no longer be removed. According to recent findings, however, amyloid deposits can at least be reduced by removing the precursor proteins.
The amyloid deposits destroy the architecture of the organs and thus lead to functional disorders. There is evidence that the deposits also exert a direct toxic effect on cells.
Diagnosis
The symptoms of amyloidosis are often non-specific, especially in the early stages of the disease. For this reason, the diagnosis is often only made in an advanced stage of the disease. Amyloidosis should be considered in the presence of otherwise unexplained proteinuria, as well as in patients with cardiomyopathy, neuropathy, liver enlargement, or multiple myeloma.
Since, if scintigraphic evidence of cardiac ATTR amyloidosis (transthyretin amyloidosis) has not been obtained when a monoclonal gammopathy has been excluded, the exact composition of the amyloid must be known for treatment, a biopsy (tissue sample) for histological amyloid detection with subtyping is usually required. This can be obtained from an affected organ (kidney, heart, stomach). A recent discovery is that generalized amyloidoses can also be classified with biopsies from subcutaneous adipose tissue, a less stressful procedure for the patient. Other less burdensome sampling sites include the minor salivary glands, gums, rectum, or skin. The histologically prepared specimens are stained with Congo red and examined by immunohistochemistry. Amyloid binds the dye Congo red and then becomes visible with a greenish glow under polarized light. The electron microscope shows that the amyloid deposits consist of fibrils, irregularly arranged, thread-like structures of different lengths with a diameter between 8 and 15 nm.
If amyloidosis has been confirmed by tissue sampling, a search for light chain disease should be performed by bone marrow aspiration, blood and urine tests. In the bone marrow, a monoclonal proliferation of plasma cells, i.e. plasma cells that produce either only kappa or only lambda chains, is indicative of AL amyloidosis. The light chains can also be detected by immunoelectrophoresis in serum or urine.
The extent of amyloid deposition can be visualized by scintigraphy with radiolabeled substances that bind to amyloid. Binding to amyloid are technetium Tc 99m-pyrophosphate, technetium-labeled aprotinin, and  I-labeled serum amyloid P component. The extent of cardiac involvement can be visualized by echocardiography or magnetic resonance imaging.
I-labeled serum amyloid P component. The extent of cardiac involvement can be visualized by echocardiography or magnetic resonance imaging.
In many cases, the diagnosis is made only after death by autopsy. An oversized tongue and characteristic coarse subcutaneous swellings point the pathologist to the correct diagnosis. Because of the bacon-like appearance of the cut surfaces of affected organs, they are also called bacon liver, bacon spleen, or bacon kidney. Just like Virchow, he can sprinkle the cut surface of any organ (so also tongue, heart) with Lugol's solution, which, with amyloid, gives a characteristic brown colour.
Related articles
Author
AlegsaOnline.com Amyloidosis: causes, types, diagnosis, and clinical significance Leandro Alegsa
URL: https://en.alegsaonline.com/art/3704
Sources
- doi.org : 10.1016/S0140-6736(15)01274-X
- worldcat.org : 1474-547X
- pubmed.ncbi.nlm.nih.gov : 26719234
- doi.org : 10.1146/annurev.med.57.121304.131243
- worldcat.org : 0066-4219
- pubmed.ncbi.nlm.nih.gov : 16409147
- oxfordmedicine.com : "Oxford Textbook of Medicine"
- doi.org : 10.1093/med/9780199204854.001.1/med-9780199204854
- doi.org : 10.3109/13506129.2014.964858
- worldcat.org : 1744-2818
- pubmed.ncbi.nlm.nih.gov : 25263598