Cystic fibrosis: causes, symptoms, diagnosis, and management
Cystic fibrosis is an inherited disorder caused by mutations in the CFTR gene. It primarily affects the lungs and digestive system and is managed with airway clearance, medications, nutrition, and specialized therapies.
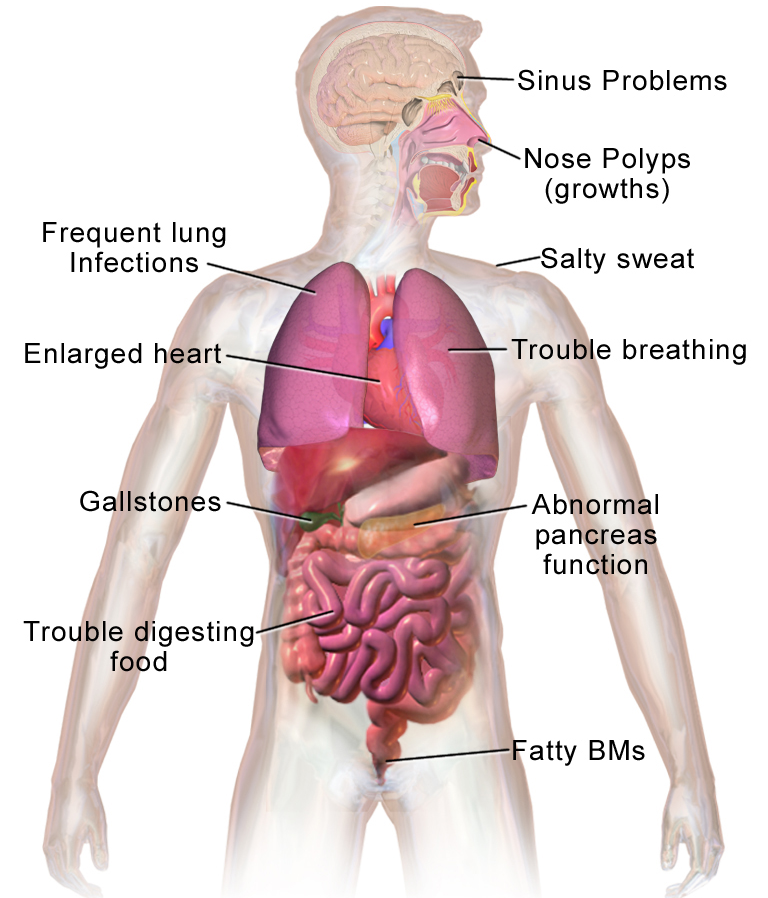
Cystic fibrosis (CF), also called mucoviscidosis, is an inherited condition that alters how certain epithelial cells handle salt and water. The result is thick, sticky secretions that interfere with normal organ function. These secretions most visibly accumulate in the airways and the digestive tract, but CF can affect multiple organs and systems throughout life. The condition is genetic and lifelong; while there is no universal cure, many treatments reduce complications and improve quality of life.
Image gallery
4 Images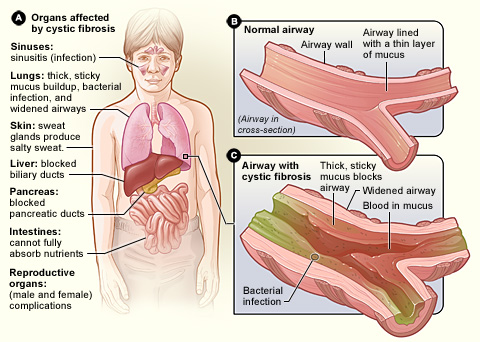

Characteristics and common effects
The core problem of CF is abnormal transport of chloride and sodium across the surface of cells, producing unusually viscous mucus. This mucus:
- Clogs airways and promotes chronic lung infections and inflammation, leading to cough, wheeze and progressive breathing difficulties.
- Blocks pancreatic ducts, impairing digestion and nutrient absorption and often causing poor weight gain despite good appetite.
- Can obstruct other organs, interfere with male fertility through absence or blockage of the vas deferens, and increase salt loss in sweat.
Genetics, inheritance, and diagnosis
Cystic fibrosis is inherited in an autosomal recessive pattern: a child develops CF when they inherit two defective copies of the CFTR gene, one from each parent. Carriers who have a single altered copy are usually healthy but can pass the variant to offspring. Diagnosis commonly begins with newborn screening in many countries, followed by confirmatory tests such as genetic testing and the sweat chloride test, which measures the concentration of salt in sweat. Clinical evaluation includes lung function testing and assessment of pancreatic status.
Treatment and long-term management
Care for people with CF is multidisciplinary and tailored to symptoms and complications. Core elements of management include:
- Regular airway clearance techniques and physiotherapy to remove mucus from the lungs.
- Inhaled and oral medications: bronchodilators, mucolytics, and antibiotics to control infection and reduce inflammation. Learn more about common treatments here.
- Nutritional support and pancreatic enzyme replacement to aid digestion and maintain healthy weight.
- Targeted CFTR modulator therapies that correct or improve the faulty protein’s function for some genetic variants.
- Advanced care options, including intravenous antibiotics for severe infections and, in selected cases, lung transplantation.
History, research, and recent advances
Cystic fibrosis was first characterized as a distinct disease in the early 20th century and more fully described by pediatricians in the 1930s. The gene responsible (CFTR) was identified in 1989, a milestone that opened the door to genetic testing and the development of precision medicines. Over recent decades, newborn screening, aggressive infection control, nutritional care and new drug classes have markedly improved outcomes and life expectancy for many people with CF. Research continues into gene therapies, improved modulators and ways to prevent chronic lung damage.
Important distinctions and practical notes
CF is not contagious — it cannot be transmitted between people. Being a carrier is different from having the disease: carriers usually have no symptoms but can pass the altered gene to children. Because CF affects organs beyond the lungs, care teams often include pulmonologists, gastroenterologists, dietitians and physiotherapists. For more on how CF affects the lungs, pancreas and other systems see lung effects, digestive complications and general information about abnormal mucus here. Genetic counseling resources and explanations about inheritance are available here.
Epidemiology
In Europe, the probability of a child being born with cystic fibrosis is about 1:2,000. In Germany, there are currently about 8,000 people living with cystic fibrosis. Worldwide, there are about 70,000, of which 30,000 are in Europe and 30,000 in North America. About 300 children with cystic fibrosis are born in Germany every year. Cystic fibrosis was at the top of the list of hereditary diseases that used to cause death in children up to the age of 15.
| The most frequent CFTR mutations in D-A-CH | ||||||
| Mutation | Gene segment | Mutation type | Class | D [%] | A [%] | CH [%] |
| ΔF508 | exon 10 | Amino acid deletion | II | 71,8 | 62,9 | 64,1 |
| R553X | exon 11 | Stop mutation | I | 2,0 | 1,7 | 3,5 |
| N1303K | exon 21 | Amino acid substitution | II | 1,8 | 0,6 | 1,6 |
| R347P | exon 7 | Amino acid substitution | IV | 1,2 | 1,6 | 0,8 |
| G542X | exon 11 | Stop mutation | I | 1,2 | 3,3 | 1,6 |
| G551D | exon 11 | Amino acid substitution | III | 0,9 | 1,2 | 0,2 |
| 1717 1G→A | Intron 10 | Splice mutation | I | 0,9 | 0,8 | 3,8 |
| 2789 +5G→A | Intron 14b | Splice mutation | IV | 0,9 | 2,4 | 0,3 |
| 3905insT | exon 20 | Frameshift mutation | I | - | - | 4,8 |
The allele frequency in the German population is about 0.02 to 0.025. This number is a measure of the relative frequency of an allele in a population. It follows that about 4% of the population, i.e. every twenty-fifth person, carries a defective CFTR gene. These approximately three million people in Germany alone are (with exceptions) healthy gene carriers who can pass on the mutated allele. In this case, they are referred to as heterozygous trait carriers. The probability of two heterozygous trait carriers fathering a child - in relation to the total population - then has a probability of 0.02² = 0.0004. With a population of 81.2 million people (as of December 2014, Federal Statistical Office), this would correspond to a mathematical 32,000 inhabitants. In epidemiological studies, a value of 1:3,300 was determined for Germany. Globally, the incidence of cystic fibrosis varies greatly. Worldwide, Ireland has the highest probability of giving birth to a child with cystic fibrosis with 1:1,800. The lowest value in Europe is found in Finland with 1:25,000. People of African descent have a risk of about 1 in 17,000, while people of Asian descent are least likely to be born with the condition, at about 1 in 100,000. For example, the figure in Japan is 1 in 350,000.
Currently, different mutations in the CFTR gene are statistically recorded in 2019 (as of June 2017). These mutations are unevenly distributed across the total population. For example, fewer than 20 mutations account for more than 0.1% and only five mutations account for more than 1% of the total number of cystic fibrosis cases. By far the most common mutation has the designation ΔF508. It is found in about 2/3 of all CFTR alleles of cystic fibrosis patients. Within Europe, the prevalence decreases from north-west to south-east.
Among the mutations recorded in 2019, non-synonymous mutations, i.e. point mutations in which a different amino acid is coded for, account for the majority with 39.4 %. This is followed by frameshift mutations, which are shifts in the reading frame of CFTR on DNA, at 15.7% and splicing mutations at 11.3%. Nonsense mutations account for 8.4 %, and in frame deletions and in frame insertions for 2.1 %. Large insertions and deletions have a frequency of 2.6%. Promoter mutations in the CFTR gene, which are point mutations in the promoter region of CFTR resulting in decreased gene expression of CFTR, are comparatively rare with a proportion of 0.74 %. Only 13.3% of the detected mutations in coding regions of CFTR do not lead to disease. In 6.5 %, the type of mutation is still unknown.
Due to the founder effect, some rather rare mutations may be significantly overrepresented in some populations. The founder effect can result from religious, ethnic or geographic isolation. For example, the stop mutation W1282X occurs in Ashkenazi Jews, the deletion 394delTT in Nordic peoples, the insertion 3905InsT in Switzerland, the amino acid substitution S549R in Bedouins, and the splice mutation 3120+1G→A on the African continent occur comparatively frequently in cystic fibrosis patients. A special case is the mutation 3905InsT. It is only common in Switzerland, in the Amish community in North America and in Acadians. In Switzerland, it ranks second among CFTR mutations with a frequency of 4.8%. Among the Amish it even reaches a value of 16.7 % and among Acadians 14.3 %. The reason for this is the founder effect of emigrants from German-speaking Switzerland, who founded the Amish community in the 18th century and also settled in Louisiana. In Germany, the ΔF508 mutation is found in 72 % of patients, but the remaining 28 % exhibit a markedly heterogeneous and diverse spectrum, which complicates genetic diagnosis. More than 80 different mutations have been detected in German cystic fibrosis patients to date. Since the mutation form influences the course of the disease and increasingly also the treatment options, the clinical prognosis for comparatively rare mutation forms in the CFTR gene is often difficult to establish. There are too few patients and thus hardly any case studies with the same CFTR genotype. Of the mutations known in 1995, about one in three is so rare that it has only been found in a single family (private mutation). Spontaneous mutations are extremely rare. Only four cases have been described worldwide to date.
Genetics and Molecular Biology
Cystic fibrosis is caused by various mutations in the CFTR gene, which in humans is located on the long arm of chromosome 7 (gene locus q31.2). The CFTR gene codes for the protein Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). This gene product functions as a chloride channel in the cell membrane. The alteration in the gene also alters the protein, and the channel function is absent or impaired. This is therefore a mutation that leads to a loss of function of the affected protein (loss-of-function mutation). The most common mutation of this gene is called ΔF508. ΔF508 refers to the absence of the amino acid phenylalanine ('F' in the one-letter code) at position 508 in the CFTR protein and affects about seven out of ten people with cystic fibrosis.
So far, more than 2000 different mutations of the CFTR gene are known, which occur more or less frequently in different populations. A special feature is that in cystic fibrosis two different mutations of the CFTR gene, i.e. two different alleles of the same gene, can nevertheless lead to the disease. This special constellation of autosomal recessive inheritance is accordingly called compound heterozygosity.
Since cystic fibrosis is inherited in an autosomal recessive manner, the disease only occurs if the trait carrier inherits one mutated gene from each parent. If both parents are carriers of one mutated and one unmodified gene each, the probability that a child will receive two intact gene copies is 25%. The probability that the child with one intact and one mutated copy is healthy but can pass on the mutation is 50%, and the probability that the child will become ill, i.e. inherit the disease-causing variant from both parents, is also 25%. If both parents have the disease, all children would also inherit the disease. However, this is very unlikely, as the affected persons are usually infertile.

Questions and answers
Q: What is cystic fibrosis?
A: Cystic fibrosis is a condition that causes the body to produce thick, sticky mucus that can build up in the lungs, digestive system, and other parts of the body.
Q: How is cystic fibrosis caused?
A: Cystic fibrosis is caused by inheriting the cystic fibrosis gene from both parents.
Q: Can a person with just one cystic fibrosis gene pass the condition down to their child?
A: A person with only one cystic fibrosis gene might not have the condition themselves but can still pass the gene onto their child.
Q: Is cystic fibrosis contagious?
A: No, cystic fibrosis is not contagious and cannot be passed from one person to another.
Q: Is there a cure for cystic fibrosis?
A: No, there is currently no cure for cystic fibrosis, but there are many medications that can help manage the condition.
Q: What parts of the body can be affected by cystic fibrosis?
A: Cystic fibrosis can affect the lungs, the digestive system, and other parts of the body.
Q: How can people with cystic fibrosis stay healthy?
A: People with cystic fibrosis can stay healthy by taking medication, monitoring their condition, and following a healthy lifestyle.
Related articles
Author
AlegsaOnline.com Cystic fibrosis: causes, symptoms, diagnosis, and management Leandro Alegsa
URL: https://en.alegsaonline.com/art/24952