Cardiomyopathy: types, causes, diagnosis, and treatment
Cardiomyopathy describes diseases of the heart muscle that reduce cardiac function. This article explains types, causes, diagnosis, therapies, prognosis, and notable distinctions.
Overview
Cardiomyopathy is a general term for disorders that primarily affect the myocardium, the muscle of the heart. When the heart muscle becomes weak, thickened, rigid, or otherwise abnormal, its ability to pump and fill with blood is impaired. People with cardiomyopathy are frequently at increased risk of arrhythmia and, in some cases, sudden cardiac death. The clinical picture ranges from silent disease discovered incidentally to progressive heart failure or life‑threatening rhythm disturbances.
Image gallery
4 Images
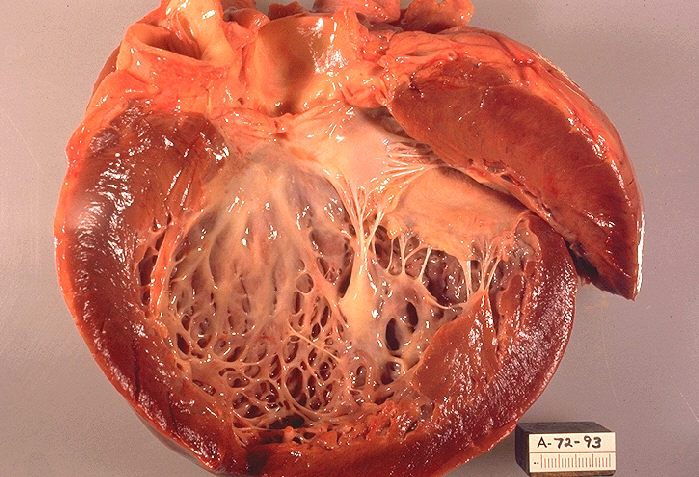


Types and characteristic features
Clinicians classify cardiomyopathies by their predominant structural or functional pattern. The principal categories include:
- Dilated cardiomyopathy — enlargement of one or both ventricles with reduced pumping function.
- Hypertrophic cardiomyopathy — thickening of the heart muscle, often the septum, which can obstruct outflow and disturb relaxation.
- Restrictive cardiomyopathy — stiff ventricles that resist filling despite relatively preserved contraction.
- Arrhythmogenic cardiomyopathy — progressive replacement of heart muscle by fibrous or fatty tissue, often affecting the right ventricle and predisposing to arrhythmias.
Other recognized forms include stress‑related (takotsubo) cardiomyopathy, infiltrative types (for example due to amyloid), and myocarditis‑related dysfunction. Some cases are labelled primary (originating in the heart) and others secondary to systemic disease.
Causes, presentation, and diagnosis
Causes vary: inherited genetic mutations, viral infections, autoimmune processes, toxins (including alcohol and some chemotherapy agents), metabolic disorders, and pregnancy can all play a role. Symptoms commonly include breathlessness on exertion, fatigue, swelling of the legs, palpitations, chest discomfort, or fainting. Physical findings and symptoms overlap with other cardiac conditions, so evaluation typically combines electrocardiography, echocardiography, cardiac magnetic resonance imaging, blood tests, and sometimes endomyocardial biopsy or genetic testing to define cause and extent.
Management and prognosis
Treatment aims to relieve symptoms, prevent progression, and reduce the risk of arrhythmia and sudden death. Standard measures include medications (such as agents to reduce workload on the heart, control rhythm, or remove excess fluid), device therapy (implantable cardioverter‑defibrillators, pacemakers, or cardiac resynchronization), catheter procedures for arrhythmia control, and mechanical circulatory support for advanced failure. In refractory cases, heart transplantation may be considered. Prognosis is highly variable and depends on the type, severity, underlying cause, and response to therapy; family screening and lifestyle modifications are often recommended.
History, research, and important distinctions
The concept of cardiomyopathy emerged as clinicians recognized heart muscle disorders not explained by coronary artery disease or valvular defects. Advances in imaging, molecular genetics, and cellular biology have clarified many inherited forms and helped develop targeted care. Important distinctions include cardiomyopathy versus ischemic heart disease (where narrowed arteries cause muscle damage) and primary versus secondary forms. Because some types carry a genetic risk, relatives of affected individuals may be offered evaluation and counseling.
Notable facts
- Cardiomyopathy is a leading cause of heart transplantation and a frequent contributor to chronic heart failure.
- Risk of dangerous arrhythmias makes prompt diagnosis and individualized risk assessment central to care; devices such as an implantable cardioverter‑defibrillator can be life‑saving in selected patients.
- Ongoing research focuses on gene‑based diagnostics, novel pharmacologic targets, and improved mechanical support technologies.
History
In the mid-1800s, chronic myocarditis alone was known as myocardial disease. Around 1900, the term primary myocardial disease was coined, and it was not until 1957 that the term cardiomyopathy emerged. There were several definitions until 1980, when the WHO referred to cardiomyopathy as "myocardial disease of unknown cause". The 1995 WHO classification expanded the term to "cardiomyopathies that result in dysfunction of the heart." New diseases such as arrhythmogenic right ventricular and restrictive cardiomyopathy were included.
Primary cardiomyopathies
Congenital primary cardiomyopathies
hypertrophic cardiomyopathy
→ Main article Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is characterized by a usually asymmetric thickening of the muscles of the left ventricle.
Arrhythmogenic right ventricular cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy (ARVCM), formerly also called arrhythmogenic right ventricular dysplasia (ARVD), is a predominantly congenital disease. In Veneto, the prevalence is particularly high, ranging from 1:2000 to 1:5000. However, there are also cases of ARVCM in Germany. As the disease progresses, more and more musculature of the right ventricle is replaced by fatty tissue, causing the right ventricle to enlarge. Rarely, restrictions in the pumping function of the heart are found. More common is sudden cardiac death (PHT) or "near" PHT triggered by physical exertion, such as competitive sports, especially in young people. Diagnosis can be made by echocardiography, MRI, ECG and McKenna score. A cardioverter defibrillator may be implanted for treatment. Athletic exertion should be avoided. Heart transplantation is the last resort in many advanced cases. Since ARVCM is inherited in many cases, it is advisable to have the family members of those affected tested. In some countries, for example Italy and the United States, all members of sports clubs are prophylactically screened.
One cause of arrhythmogenic right ventricular cardiomyopathy may be mutations in desmosome proteins. Desmosomes are important for cell contact of cells. In the case of ARVCM specifically for the myocardium. In Naxos disease, for example, the DSP gene is affected by a mutation that codes for the cell adhesion protein desmoplakin. The gene defect leads to arrhythmogenic right ventricular cardiomyopathy in affected patients. Mutations in the DES gene, which codes for the intermediate filament desmin, can also lead to ARVCM.
Left ventricular hypertrabeculation
Congenital, rare myocardial disease with spongy distended muscles, especially in the apex of the left ventricle, showing deep hollows (sinusoids) between muscle fibers (trabeculae) connected to the cardiac cavity. In isolated noncompaction of the myocardium (syn: noncompaction cardiomyopathy, left ventricular hypertrabeculation, spongy myocardium), the myocardium has not further compacted from its loose meshwork during the early embryonic period (spongy myocardium). Common in skeletal muscle diseases, also in combination with complex cyanotic heart defects.
The diagnosis is made by echocardiography, MRI or angiography of the left ventricle during cardiac catheterization. The clinical course is unclear. Cases of severe heart failure, thromboembolism, arrhythmias and sudden cardiac death have been reported. Familial clustered cases have been described, with mutations of the Z-disk, mitochondrial and G4.5 gene for tafazzin isolated.
Glycogen storage diseases
A distinction is made between PRKAG2 and Danon, a type II glycogenosis.
Line defects
Lenègre disease is a primary progressive conduction defect of the His-Purkinje system leading to widening of the QRS complex on the ECG with long pauses, bradycardia and syncope.
Sick sinus syndrome phenotypically resembles a conduction defect and may be autosomal dominant.
Wolff-Parkinson-White syndrome (WPW) also rarely occurs in families.
Mitochondrial myopathies
In part, the enzymes of the respiratory chain are encoded on the genes of the mitochondria. Several syndromes due to corresponding gene defects are known, including the
- Kearns-Sayre syndrome with pigmentary degeneration of the retina of the eye, ocular muscle paralysis and cardiomyopathy, and the
- MELAS syndrome with myopathy, encephalopathy, lactic acidosis and stroke-like episodes. In addition to the four features defined by the acronym, hypertrophic cardiomyopathy and diffuse coronary vascular disease are typical. Treatment is difficult. Some patients have been treated with coenzyme Q with modest short-term success. Heart transplantation is an alternative.
Ion channel defects
There is a growing list of rare hereditary and congenital cardiac arrhythmias encoded by genes for defective ion channel proteins. Also, a small proportion of 5-10% of children with suddeninfant death syndrome might be caused by ion channel defects. Clinical diagnosis of an ion channel defect is often possible phenotypically by a standard 12-lead ECG. Some of these cases have previously been classified as idiopathic ventricular fibrillation.
Long QT syndrome (LQTS)
Long QT syndrome is probably the most common of the ion channel diseases. Characteristic are prolongations of the ventricular depolarization and the QT interval in the 12-lead ECG. A special form of ventricular tachycardia (torsade de pointes) occurs and with it a risk of syncope and sudden cardiac death.
Jervell and Lange-Nielsen syndrome: rare, autosomal recessive, associated with deafness. Two genes encoding a slow activating delayed potassium channel.
Romano-Ward syndrome: Much more common, autosomal dominant, thirteen different genes, five of which (KCNQ1, KCNH2, KCNE1, KCNE2, KCNJ2) code for various potassium channels (I(K)), two (SCN5A, SCN3B) for cardiac sodium channels (I(Na)), and one (ANK2) for the protein ankyrin B, which is responsible for anchoring ion channels in the cell membrane, among others.
Brugada Syndrome
→ Main article: Brugada syndrome
As a clinical entity, Brugada syndrome has been known since 1992, it is sometimes called Brugada-Brugada syndrome after the two brothers who first described it, Pedro and Josep Brugada. It is sometimes responsible for sudden cardiac death, especially in young people. It is characterized by right bundle-branch block-like changes in the ECG, which may be provoked by an ajmaline test. Various genetic defects are known, they were described by Ramon Brugada, the youngest of the three Brugada brothers.
Asian SUNDS
SUNDS = "sudden unexplained nocturnal death syndrome". Predominantly in young Asian men, especially from Thailand, Japan, the Philippines and Cambodia. Sudden death during sleep due to ventricular tachycardia or ventricular fibrillation. Some cases are indistinguishable in appearance from Brugada syndrome. SUNDS made its way into the "1000 Ways to Bite the Dust" series as Death #818, entitled Fatal Dream.
Short QT Syndrome (SQTS)
First described in 2000. The QT interval in the ECG is shortened to less than 330 ms. There is a great risk of sudden cardiac death.
CPVT
CPVT = "Catecholaminergic Polymorphic Ventricular Tachycardia", first described by Coumel in 1978, characterized by a striking widening of the QRS complex in the ECG as well as by syncope, polymorphic ventricular tachycardia triggered by physical exertion or by violent emotional movements in children and adolescents, and by an increased risk of sudden cardiac death. CPVT is caused by mutations in the gene RYR2, which encodes the cardiac (=subtype 2) ryanodine receptor that is predominantly expressed in cardiac muscle. The inheritance of the mutation in the RYR2 gene within the affected families follows an autosomal dominant inheritance pattern, i.e. the risk for a first-degree relative to also inherit the disease-causing mutation is 50%. In 30% of diagnosed cases of CPVT, a previous sudden cardiac death is observed in family members.
Mixed (congenital and acquired) primary cardiomyopathies
dilated cardiomyopathy
Dilated cardiomyopathy (DCM), in which initially the left ventricle (heart chamber) (in the final stage also all heart cavities) is considerably dilated (the heart can be compared to a large flaccid sac). The wall thicknesses are usually not thickened or only slightly thickened (hypertrophied). The heart contracts only to a limited extent (= systolic functional restriction), often combined with asynchronous contraction of the chambers, due to a disturbance in conduction as a result of left bundle branch block. In terms of numbers, expired myocarditis and chronic alcohol abuse are the most frequent causes. There are also congenital forms. Secondary forms include "ischemic DCM" due to coronary artery disease and the end-state of a hypertensive heart. DCM is a common reason for heart transplantation when the patient's condition cannot be adequately improved with medications, coronary intervention, or cardiac resynchronization therapy (CRT). Diagnosis is confirmed by imaging (echocardiography, MRI, MSCT) and fine tissue (myocardial biopsy) after clinical suspicion with typical symptoms. Coronary artery disease must be ruled out by cardiac catheterization, as this could result in a curative treatment option for the cause.
Restrictive cardiomyopathy
Restrictive cardiomyopathy (RCM) presents with normal sized ventricles and mostly normal systolic pump function. Due to increased incorporation of connective tissue into the cardiac musculature, the cardiac musculature hardens. The resulting stiffening of the ventricles makes it difficult for the heart to fill during the slackening phase (diastole), and the blood backs up in the atria, which are greatly enlarged as a result. The wall thickness of the left ventricle is normal and the heart valves are regular.
Patients become conspicuous by symptoms of heart failure such as dyspnea and leg edema. The disease is extremely rare in industrialized countries and can be overlooked if it is not specifically looked for. The main cause is amyloidosis; sporadic and familial forms are known. For example, mutations in the DES gene, which encodes the intermediate filament protein desmin, have been identified. In tropical countries, however, restrictive cardiomyopathy is far more common, causing up to 20% of all cardiovascular deaths. Diagnostic methods include echocardiography, with tissue Doppler if necessary, cardiac catheterization with hemodynamic measurement, myocardial biopsy if necessary, and MRI.
Acquired primary cardiomyopathies
Myocarditis: inflammatory cardiomyopathy
Myocarditis is an acute or chronic process that may be caused by a wide range of
- toxins, like cocaine,
- endogenous substances, e.g. interleukin-2,
- infectious pathogens such as
- Viruses, e.g. coxsackie virus, adenovirus, parvovirus B19, human herpes virus 6 (HHV6), HIV, (the involvement of the hepatitis C virus is controversial)
- Bacteria, e.g. diphtheria, meningococcus, psittacosis, streptococcus, borrelia
- Rickettsia, e.g. spotted fever, Rocky Mountain spotted fever
- Fungi, e.g. Aspergillus, Candida
- parasites, e.g. Trypanosoma cruzi (Chagas disease), toxoplasmosis
- Whipple's disease (intestinal lipodystrophy)
- autoimmune (giant cell myocarditis) or in the context of a
- Hypersensitivity reactions e.g. to drugs such as antibiotics, sulphonamides, anticonvulsants and antirheumatic drugs.
Endocardial fibroelastosis in newborns and infants is the result of intrauterine infection with mumps virus.
Stress provoked (Tako-Tsubo)
Tako-Tsubo cardiomyopathy (syn: stress cardiomyopathy, broken heart syndrome, apical ballooning) is a myocardial disease that usually occurs in postmenopausal women and frequently after emotional stress situations and appears like an acute myocardial infarction in terms of symptoms, ECG changes and laboratory findings. There is a balloon-like distension of the apex of the ventricle as in a severe anterior wall infarction, but no narrowing or occlusion of the coronary vessels is found. The cause is thought to be a stress hormone-induced, only temporary occlusion of the fine hair vessels of the coronary arteries. This leads to a temporary "stunning" of the heart muscle, which, unlike in a real heart attack, does not die (necrosis) but can recover completely. The prognosis is usually good and in a few months the heart muscle disorder is regressive. The mortality rate is about 3%. Diagnosis is made by echocardiography, cardiac catheterization and magnetic resonance imaging (MRI).
Pregnancy Cardiomyopathy
Gestational or peripartum cardiomyopathy is a rare cause of acquired dilated cardiomyopathy with systolic heart failure in pregnant women in the last trimester or up to 5 months after delivery (peripartum cardiomyopathy). The cause is unclear, an inflammatory component (myocarditis), immune activating processes and gestational hypertension are discussed as triggers. Mostly overweight pregnant women over 30 years of age are affected, who have already given birth several times and have had pre-eclampsia. About half of the patients almost recover after six months, but in individual cases progressive heart failure with death or heart transplantation may occur.
Tachycardiomyopathy
Tachycardia-induced cardiomyopathy (synonym: tachymyopathy) is a potentially reversible impairment of predominantly left ventricular pump function that occurs in the setting of a prolonged tachycardic arrhythmia, usually rapid atrial fibrillation. The diagnosis is made by echocardiography together with the ECG. Therapeutically, the heart rate is first lowered with cardiac glycosides as well as with beta-blockers. In case of beta-blocker intolerance, calcium antagonists of the verapamil or diltiazem type can also be used. If the rate is not reduced sufficiently, dronedarone or amiodarone can be used. Heart failure therapy is then started. If there is no improvement after a short time and no extensive normalization of the heart's pumping capacity after several weeks of optimal therapy, other causes must be sought.
Newborns of diabetic mothers with poor metabolic status
This transient and rare form of non-familial cardiomyopathy is observed in children whose diabetic mothers had poor metabolic status (high blood glucose levels) during pregnancy. It often occurs together with fetal macrosomia.



Questions and answers
Q: What is cardiomyopathy?
A: Cardiomyopathy is a heart muscle disease that impairs the function of the myocardium.
Q: What are the consequences of having cardiomyopathy?
A: People with cardiomyopathy are often at risk of arrhythmia and/or sudden cardiac death.
Q: How many types of cardiomyopathy are there?
A: There are four main types of cardiomyopathy.
Q: What is the reason behind cardiomyopathies?
A: The function of the myocardium is impaired for any reason in cardiomyopathies.
Q: Is there only one cause for cardiomyopathy?
A: No, there are multiple reasons for cardiomyopathy.
Q: Is cardiomyopathy a common illness?
A: Cardiomyopathy is not a common illness, but it is a dangerous one.
Q: Can people with cardiomyopathy lead a completely normal life?
A: It depends on the severity of the condition, but people with cardiomyopathy may need to take precautions and undergo treatment in order to lead a normal life.
Tags
Related articles
Author
AlegsaOnline.com Cardiomyopathy: types, causes, diagnosis, and treatment Leandro Alegsa
URL: https://en.alegsaonline.com/art/16951
Sources
- cardiomyopathy.org : "About cardiomyopathy"